

Protein disulfide isomerase as a novel target for cyclopentenone prostaglandins: implications for hypoxic ischemic injury
- PMID: 25754985
- PMCID: PMC4972022
- DOI: 10.1111/febs.13259
Protein disulfide isomerase as a novel target for cyclopentenone prostaglandins: implications for hypoxic ischemic injury
Abstract
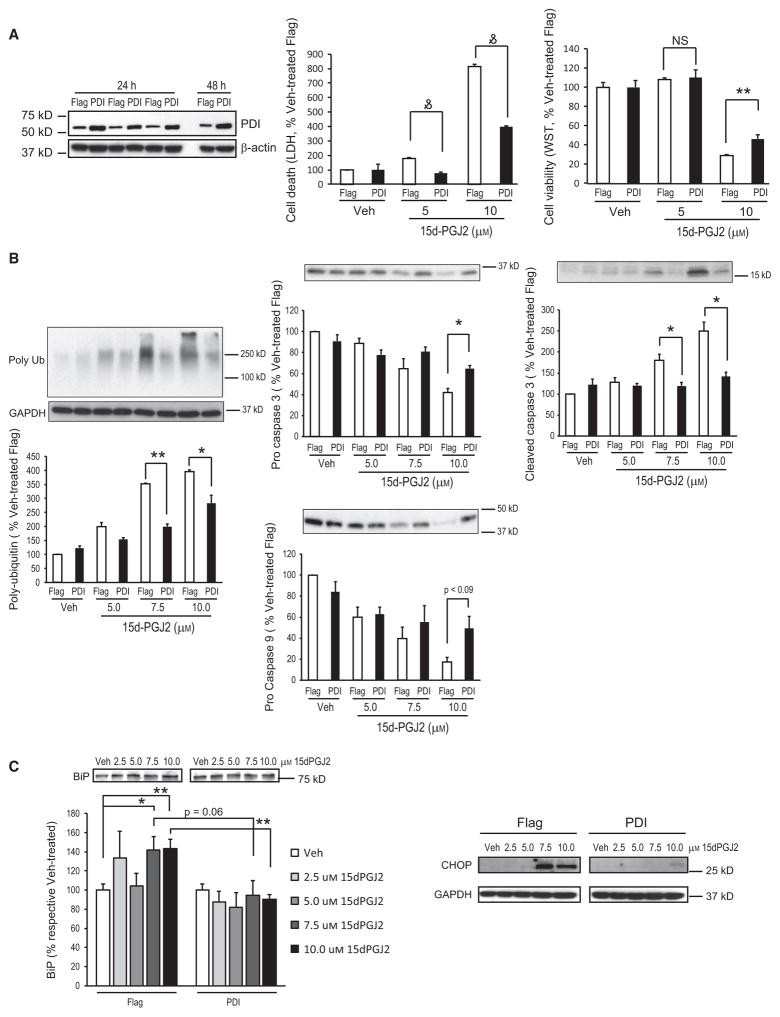
Cyclooxygenase-2 (COX-2) is an important contributor to ischemic brain injury. Identification of the downstream mediators of COX-2 toxicity may allow the development of targeted therapies. Of particular interest is the cyclopentenone family of prostaglandin metabolites. Cyclopentenone prostaglandins (CyPGs) are highly reactive molecules that form covalent bonds with cellular thiols. Protein disulfide isomerase (PDI) is an important molecule for the restoration of denatured proteins following ischemia. Because PDI has several thiols, including thiols within the active thioredoxin-like domain, we hypothesized that PDI is a target of CyPGs and that CyPG binding of PDI is detrimental. CyPG-PDI binding was detected in vitro via immunoprecipitation and MS. CyPG-PDI binding decreased PDI enzymatic activity in recombinant PDI treated with CyPG, and PDI immunoprecipitated from neuronal culture treated with CyPG or anoxia. Toxic effects of binding were demonstrated in experiments showing that: (a) pharmacologic inhibition of PDI increased cell death in anoxic neurons, (b) PDI overexpression protected neurons exposed to anoxia and SH-SY5Y cells exposed to CyPG, and (c) PDI overexpression in SH-SY5Y cells attenuated ubiquitination of proteins and decreased activation of pro-apoptotic caspases. In conclusion, CyPG production and subsequent binding of PDI is a novel and potentially important mechanism of ischemic brain injury. We show that CyPGs bind to PDI, cyclopentenones inhibit PDI activity, and CyPG-PDI binding is associated with increased neuronal susceptibility to anoxia. Additional studies are necessary to determine the relative role of CyPG-dependent inhibition of PDI activity in ischemia and other neurodegenerative disorders.
Keywords: brain; cyclooxygenase; cyclopentenone; ischemia; protein disulfide isomerase.
© 2015 FEBS.
Figures




References
-
- Candelario-Jalil E, Fiebich BL. Cyclooxygenase inhibition in ischemic brain injury. Curr Pharm Des. 2008;14:1401–1418. - PubMed
-
- Amer M, Bead VR, Bathon J, Blumenthal RS, Edwards DN. Use of nonsteroidal anti-inflammatory drugs in patients with cardiovascular disease: a cautionary tale. Cardiol Rev. 2010;18:204–212. - PubMed
-
- Iadecola C, Gorelick PB. The Janus face of cyclooxygenase-2 in ischemic stroke: shifting toward downstream targets. Stroke. 2005;36:182–185. - PubMed
-
- Hewett SJ, Bell SC, Hewett JA. Contributions of cyclooxygenase-2 to neuroplasticity and neuropathology of the central nervous system. Pharmacol Ther. 2006;112:335–357. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
