

Characterization of pulmonary function in Duchenne Muscular Dystrophy
- PMID: 25755201
- PMCID: PMC4402127
- DOI: 10.1002/ppul.23172
Characterization of pulmonary function in Duchenne Muscular Dystrophy
Abstract
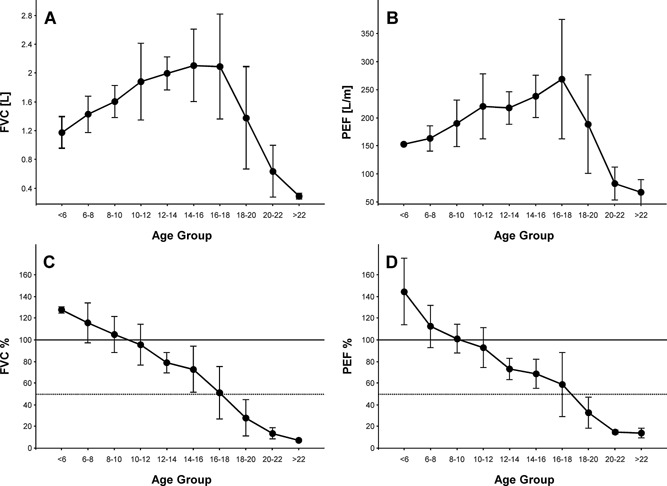
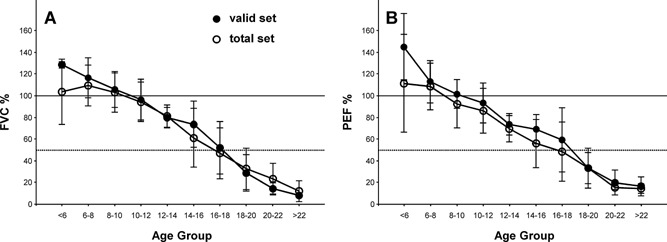
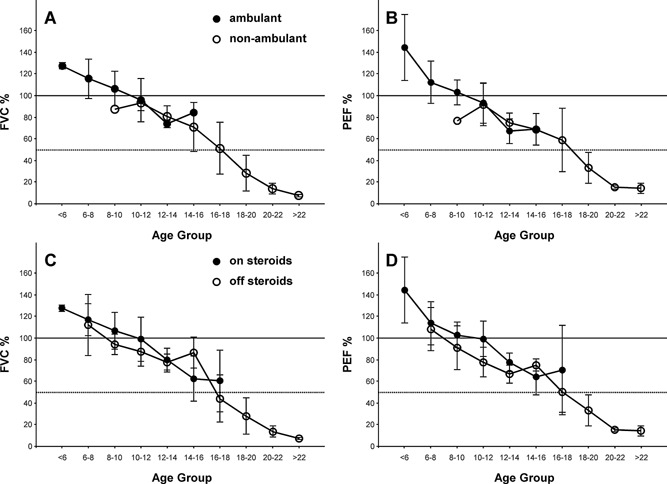
Decline in pulmonary function in Duchenne Muscular Dystrophy (DMD) contributes to significant morbidity and reduced longevity. Spirometry is a widely used and fairly easily performed technique to assess lung function, and in particular lung volume; however, the acceptability criteria from the American Thoracic Society (ATS) may be overly restrictive and inappropriate for patients with neuromuscular disease. We examined prospective spirometry data (Forced Vital Capacity [FVC] and peak expiratory flow [PEF]) from 60 DMD patients enrolled in a natural history cohort study (median age 10.3 years, range 5-24 years). Expiratory flow-volume curves were examined by a pulmonologist and the data were evaluated for acceptability using ATS criteria modified based on the capabilities of patients with neuromuscular disease. Data were then analyzed for change with age, ambulation status, and glucocorticoid use. At least one acceptable study was obtained in 44 subjects (73%), and 81 of the 131 studies (62%) were acceptable. The FVC and PEF showed similar relative changes in absolute values with increasing age, i.e., an increase through 10 years, relative stabilization from 10-18 years, and then a decrease at an older age. The percent predicted, FVC and PEF showed a near linear decline of approximately 5% points/year from ages 5 to 24. Surprisingly, no difference was observed in FVC or PEF by ambulation or steroid treatment. Acceptable spirometry can be performed on DMD patients over a broad range of ages. Using modified ATS criteria, curated spirometry data, excluding technically unacceptable data, may provide a more reliable means of determining change in lung function over time.
Keywords: Muscular dystrophy; forced vital capacity; natural history; peak expiratory flow; pulmonary function test.
© 2015 The Authors. Pediatric Pulmonology published by Wiley Periodicals, Inc.
Figures
Comment in
-
The natural history of pulmonary function in Duchenne muscular dystrophy.Pediatr Pulmonol. 2015 May;50(5):421-2. doi: 10.1002/ppul.23170. Epub 2015 Mar 3. Pediatr Pulmonol. 2015. PMID: 25736812 No abstract available.
References
-
- Emery AE. Population frequencies of inherited neuromuscular diseases‐a world survey. Neuromuscul Disord 1991; 1:19–29. - PubMed
-
- Hoffman EP, Brown RH, Kunkel LM. Dystrophin: The protein product of the duchenne muscular dystrophy locus. Cell 1987; 51:919–928. - PubMed
-
- Hoffman EP, Fischbeck KH, Brown RH, Johnson M, Medori R, Loike JD, Harris JB, Waterston R, Brooke M, Specht L. Characterization of dystrophin in muscle‐biopsy specimens from patients with Duchenne's or Becker's muscular dystrophy. N Engl J Med 1988; 318:1363–1368. - PubMed
-
- McDonald CM, Abresch RT, Carter GT, Fowler WM, Jr. , Johnson ER, Sigford BJ. Profiles of neuromuscular diseases—Duchenne muscular dystrophy. Am J Phys Med Rehabil 1995; 74:S70–S92. - PubMed
-
- Finder JD, Birnkrant D, Carl J, Farber HJ, Gozal D, Iannaccone ST, Kovesi T, Kravitz RM, Panitch H, Schramm C, et al. Respiratory care of the patient with Duchenne muscular dystrophy: ATS Consensus Statement. Am J Respir Crit Care Med 2004; 170:456–465. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
