

A review and current perspective on Wilson disease
- PMID: 25755520
- PMCID: PMC3940372
- DOI: 10.1016/j.jceh.2013.06.002
A review and current perspective on Wilson disease
Abstract
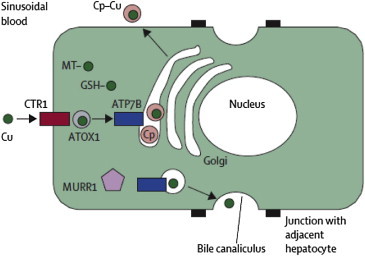
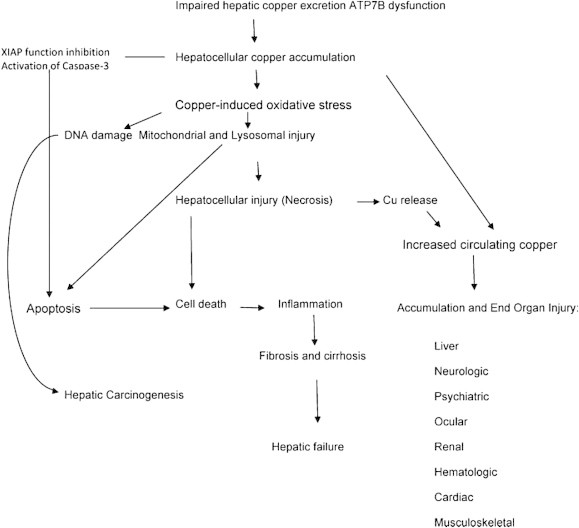
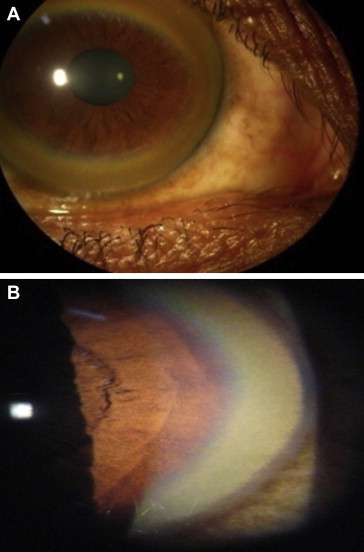
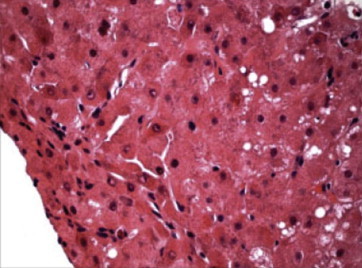
Wilson disease is a rare, inherited autosomal recessive disease of copper metabolism and may be more common where consanguinity is prevalent. Much has been known about the disease after it was first described by Kinnier Wilson as 'progressive lenticular degeneration in 1912. Over 500 mutations of the ATP7B gene has been identified with no clear genotype to phenotype correlation. Loss of ATP7B function leads various grades of reduced biliary excretion of copper and reduced incorporation of copper into ceruloplasmin; accumulation and toxicity of copper in the liver, brain and other tissues results in liver toxicity and other myriad manifestations of the disease. The clinical features may vary from asymptomatic state to chronic liver disease, acute liver failure, neuropsychiatric manifestations and hemolytic anemia. Diagnosis is based on the combination of clinical sign's, biochemical features, histologic findings and mutation analysis of ATP7B gene. Subtle geographical differences exist with a disproportionate proportion of children presenting with acute liver failure. A high index of suspicion is needed for an early diagnosis. Ratios of biochemical indices for early diagnosis need validation across geographical regions and may not be particularly applicable in children. Better biomarkers or the need for tests for early detection of ALF persists. Drugs used in the treatment of Wilson disease include copper chelating agents such as d-Penicillamine, trientine and zinc salt. Untreated Wilson disease uniformly leads to death from liver disease or severe neurological disability. Early recognition and treatment has excellent prognosis. Liver transplantation is indicated in acute liver failure and end stage liver disease. Family screening in order to detect the disorder in the first-degree relatives is warranted. This review provides an overview of different aspects of Wilson disease including geographical differences in presentations and clinical management and the limitations of currently available tests.
Keywords: ALF, acute liver failure; ATP7B; CCS1, copper chaperone for superoxide dismutase 1; CT, computerized tomography; CTR-1, copper transporter protein; MRI, magnetic resonance imaging; OLT, orthotropic liver transplantation; SOD1, superoxide dismutase; TM, tetrathiomolybdate; UNOS, United network for organ sharing; XIAP, X linked inhibitor of apoptosis; ceruloplasmin; chelators; liver failure; mutation.
Figures

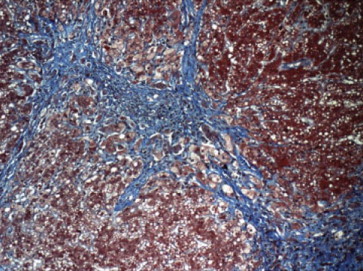
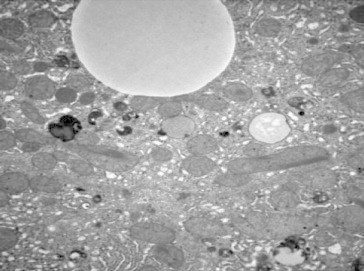
References
-
- Wilson S.A.K. Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver. Brain. 1912;34:295–507. - PubMed
-
- Bull P.C., Thomas G.R., Rommens J.M. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5:327–337. - PubMed
-
- Tanzi R.E., Petrukhin K., Chernov I. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet. 1993;5:344–350. - PubMed
-
- Yamaguchi Y., Heiny M.E., Gitlin J.D. Isolation and characterization of a human liver cDNA as a candidate gene for Wilson disease. Biochem Biophys Res Commun. 1993;197:271–277. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
