

Modeling the interaction between quinolinate and the receptor for advanced glycation end products (RAGE): relevance for early neuropathological processes
- PMID: 25757085
- PMCID: PMC4354912
- DOI: 10.1371/journal.pone.0120221
Modeling the interaction between quinolinate and the receptor for advanced glycation end products (RAGE): relevance for early neuropathological processes
Expression of concern in
-
Expression of Concern: Modeling the Interaction between Quinolinate and the Receptor for Advanced Glycation End Products (RAGE): Relevance for Early Neuropathological Processes.PLoS One. 2023 Feb 14;18(2):e0281905. doi: 10.1371/journal.pone.0281905. eCollection 2023. PLoS One. 2023. PMID: 36787334 Free PMC article. No abstract available.
Abstract
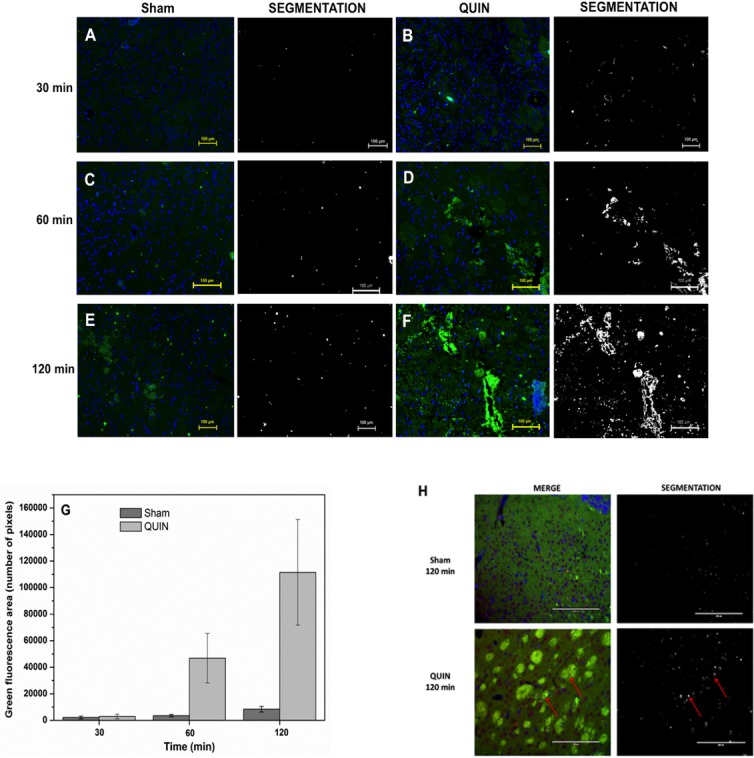
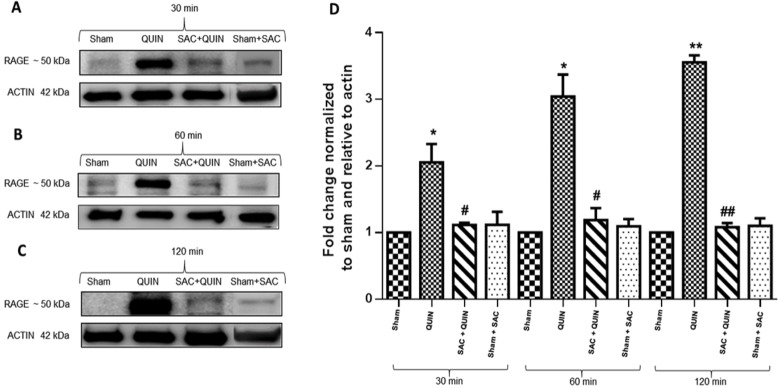
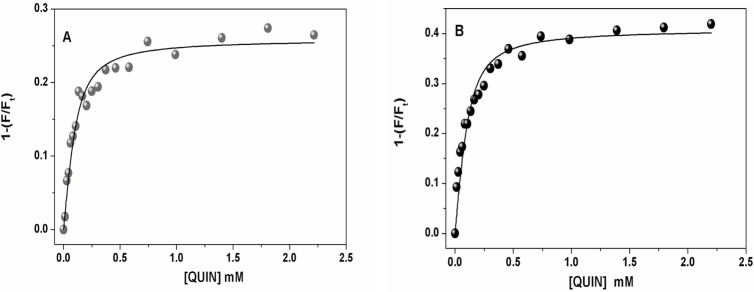
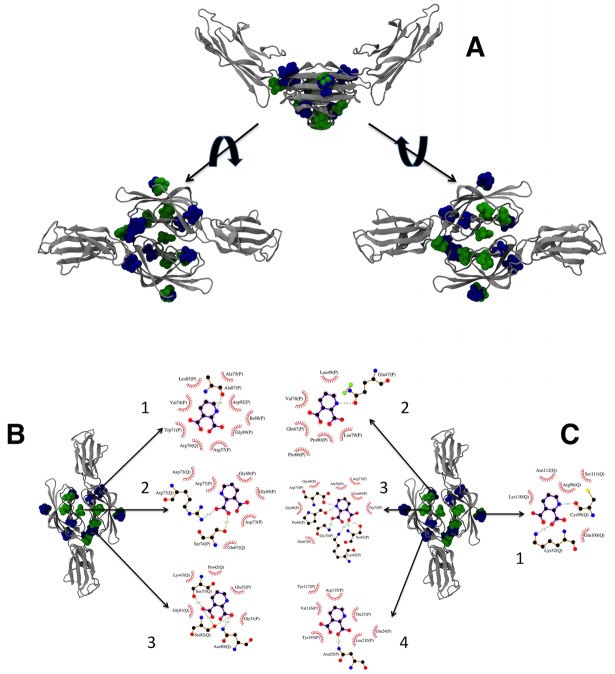
The receptor for advanced glycation end products (RAGE) is a pattern-recognition receptor involved in neurodegenerative and inflammatory disorders. RAGE induces cellular signaling upon binding to a variety of ligands. Evidence suggests that RAGE up-regulation is involved in quinolinate (QUIN)-induced toxicity. We investigated the QUIN-induced toxic events associated with early noxious responses, which might be linked to signaling cascades leading to cell death. The extent of early cellular damage caused by this receptor in the rat striatum was characterized by image processing methods. To document the direct interaction between QUIN and RAGE, we determined the binding constant (Kb) of RAGE (VC1 domain) with QUIN through a fluorescence assay. We modeled possible binding sites of QUIN to the VC1 domain for both rat and human RAGE. QUIN was found to bind at multiple sites to the VC1 dimer, each leading to particular mechanistic scenarios for the signaling evoked by QUIN binding, some of which directly alter RAGE oligomerization. This work contributes to the understanding of the phenomenon of RAGE-QUIN recognition, leading to the modulation of RAGE function.
Conflict of interest statement
Figures
References
-
- Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993; 262: 689–695. - PubMed
-
- Olney JW. Excitotoxicity: an overview. Can Dis Wkly Rep Rapp Hebd Mal Au Can 1990; 16 Suppl 1E: 47–57; discussion 57–58. - PubMed
-
- Stone TW, Mackay GM, Forrest CM, Clark CJ, Darlington LG. Tryptophan metabolites and brain disorders. Clin Chem Lab Med CCLM FESCC. 2003; 41: 852–859. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
