

Long-term follow-up of patients with phenylketonuria treated with tetrahydrobiopterin: a seven years experience
- PMID: 25757997
- PMCID: PMC4351928
- DOI: 10.1186/s13023-015-0227-8
Long-term follow-up of patients with phenylketonuria treated with tetrahydrobiopterin: a seven years experience
Abstract
Background: Phenylketonuria (PKU) is an autosomal recessive disorder caused by the deficiency of phenylalanine hydroxylase that catalyzes the conversion of phenylalanine to tyrosine, using tetrahydrobiopterin (BH4) as coenzyme. Besides dietary phenylalanine restriction, new therapeutic options are emerging, such as the treatment with BH4 in subgroups of PKU patients responding to a loading test with BH4.
Methods: A no-profit open-label interventional trial with long-term oral BH4 therapy, sponsored by the Italian Medicines Agency (AIFA), was performed in a group of 17 PKU patients resulted as BH4 responders among 46 subjects analyzed for BH4-responsiveness (prot. FARM5MATC7). We report on efficacy and safety data of BH4 therapy and analyze factors predicting BH4-responsiveness and long-term response to BH4. A BH4-withdrawal test was used as a proof of the efficacy of long-term therapy with BH4.
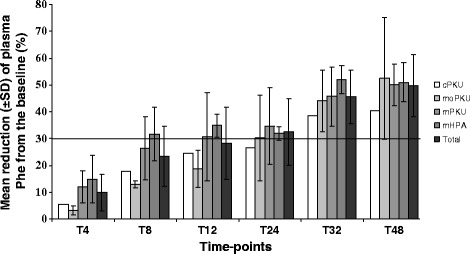
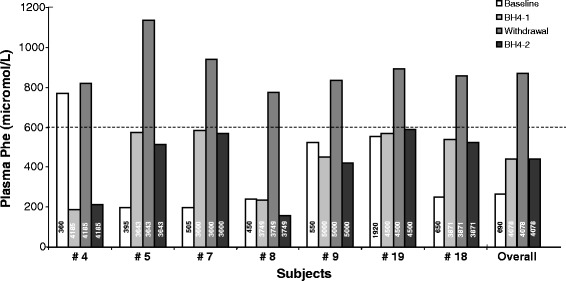
Results: Forty-four percent of the patients responded to the 48 h-long loading test with BH4. All the phenotypic classes were represented. Genotype was the best predictor of responsiveness, along with lower phenylalanine levels at diagnosis, higher tolerance and lower phenylalanine/tyrosine ratio before the test. In BH4 responder patients, long-term BH4 therapy resulted safe and effective in increasing tolerance while maintaining a good metabolic control. The BH4 withdrawal test, performed in a subset of patients, showed that improved tolerance was directly dependent on BH4 assumption. Tolerance to phenylalanine was re-evaluated in 43.5% of patients and was longitudinally analyzed in 5 patients.
Conclusions: Long-term treatment with BH4 is safe and effective in increasing tolerance to phenylalanine. There is real need to assess the actual tolerance to phenylalanine in PKU patients to ameliorate quality of life, improve nutritional status, avoiding unnecessarily restricted diets, and interpret the effects of new therapies for PKU.
Figures
References
-
- Camp KM, Parisi MA, Acosta PB, Berry GT, Bilder DA, Blau N, Bodamer OA, Brosco JP, Brown CS, Burlina AB, Burton BK, Chang CS, Coates PM, Cunningham AC, Dobrowolski SF, Ferguson JH, Franklin TD, Frazier DM, Grange DK, Greene CL, Groft SC, Harding CO, Howell RR, Huntington KL, Hyatt-Knorr HD, Jevaji IP, Levy HL, Lichter-Konecki U, Lindegren ML, Lloyd-Puryear MA, et al. Phenylketonuria Scientific Review Conference: state of the science and future research needs. Mol Genet Metab. 2014;112:87–122. doi: 10.1016/j.ymgme.2014.02.013. - DOI - PubMed
-
- Scriver CR, Kaufman S. Hyperphenylalaninemia: Phenylalanine hydroxylase deficiency. In: Scriver CR, Kaufman S, Eisensmith RC, Woo SLC, editors. The Metabolic and Molecular Bases of Inherited Disease. 8. New York: McGraw-Hill; 2003. pp. 1667–724.
-
- Kono K, Okano Y, Nakayama K, Hase Y, Minamikawa S, Ozawa N, Yokote H, Inoue Y. Diffusion-weighted MR imaging in patients with phenylketonuria: relationship between serum phenylalanine levels and ADC values in cerebral white matter. Radiology. 2005;236:630–6. doi: 10.1148/radiol.2362040611. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
