

Excision of Expanded GAA Repeats Alleviates the Molecular Phenotype of Friedreich's Ataxia
- PMID: 25758173
- PMCID: PMC4817761
- DOI: 10.1038/mt.2015.41
Excision of Expanded GAA Repeats Alleviates the Molecular Phenotype of Friedreich's Ataxia
Abstract
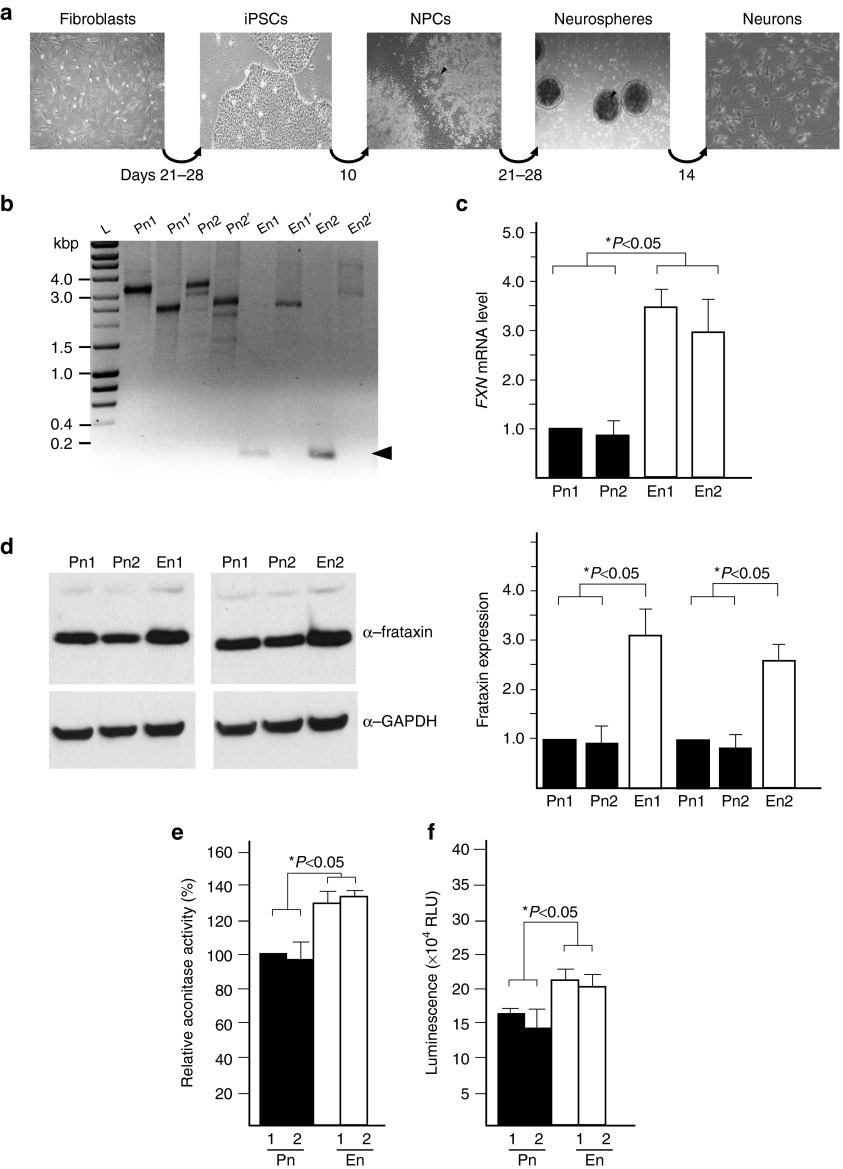
Friedreich's ataxia (FRDA) is an autosomal recessive neurological disease caused by expansions of guanine-adenine-adenine (GAA) repeats in intron 1 of the frataxin (FXN) gene. The expansion results in significantly decreased frataxin expression. We report that human FRDA cells can be corrected by zinc finger nuclease-mediated excision of the expanded GAA repeats. Editing of a single expanded GAA allele created heterozygous, FRDA carrier-like cells and significantly increased frataxin expression. This correction persisted during reprogramming of zinc finger nuclease-edited fibroblasts to induced pluripotent stem cells and subsequent differentiation into neurons. The expression of FRDA biomarkers was normalized in corrected patient cells and disease-associated phenotypes, such as decreases in aconitase activity and intracellular ATP levels, were reversed in zinc finger nuclease corrected neuronal cells. Genetically and phenotypically corrected patient cells represent not only a preferred disease-relevant model system to study pathogenic mechanisms, but also a critical step towards development of cell replacement therapy.
Figures
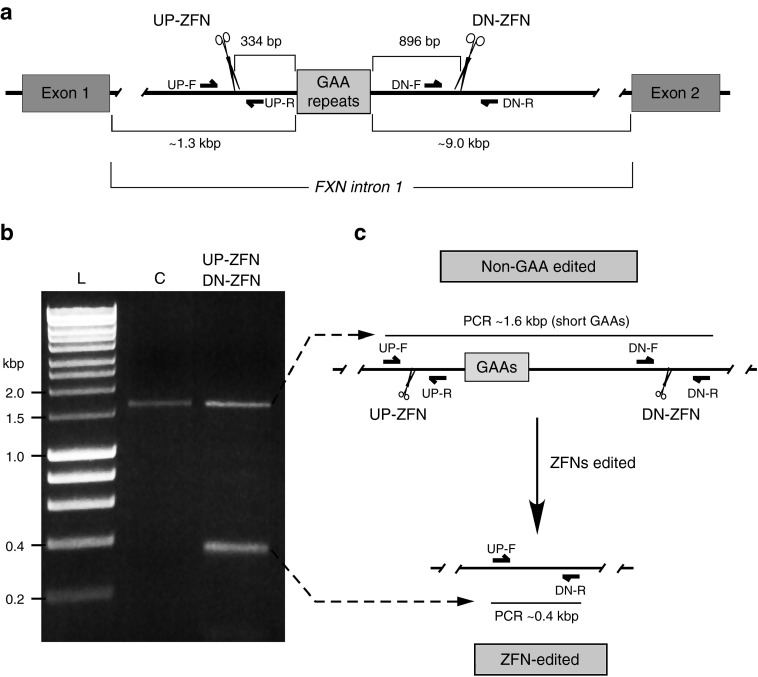
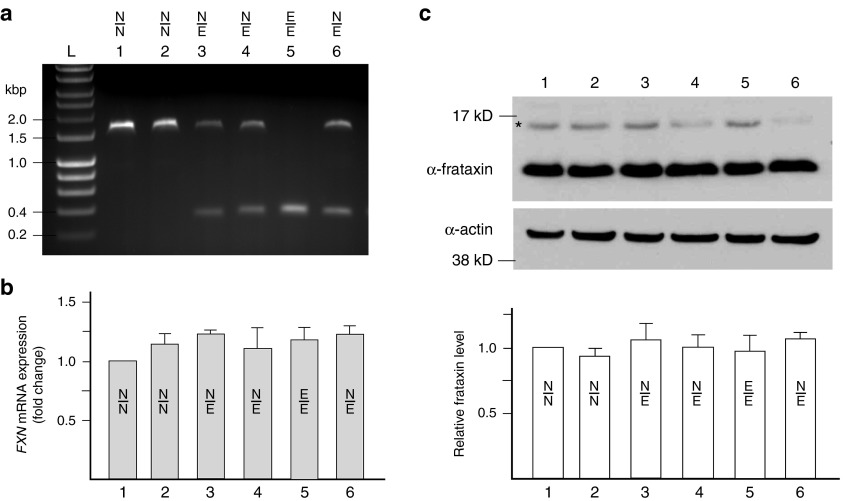
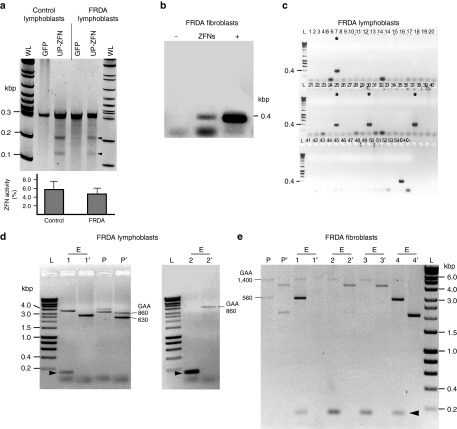
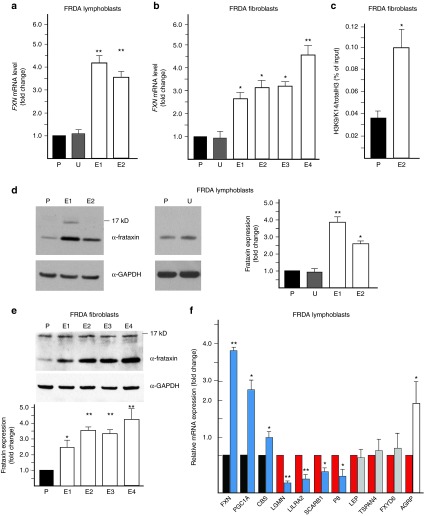
References
-
- Campuzano, V, Montermini, L, Moltò, MD, Pianese, L, Cossée, M, Cavalcanti, F et al. (1996). Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271: 1423–1427. - PubMed
-
- Herman, D, Jenssen, K, Burnett, R, Soragni, E, Perlman, SL and Gottesfeld, JM (2006). Histone deacetylase inhibitors reverse gene silencing in Friedreich's ataxia. Nat Chem Biol 2: 551–558. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
