

Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma
- PMID: 25759019
- PMCID: PMC5675004
- DOI: 10.1016/j.ccell.2015.02.001
Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma
Abstract
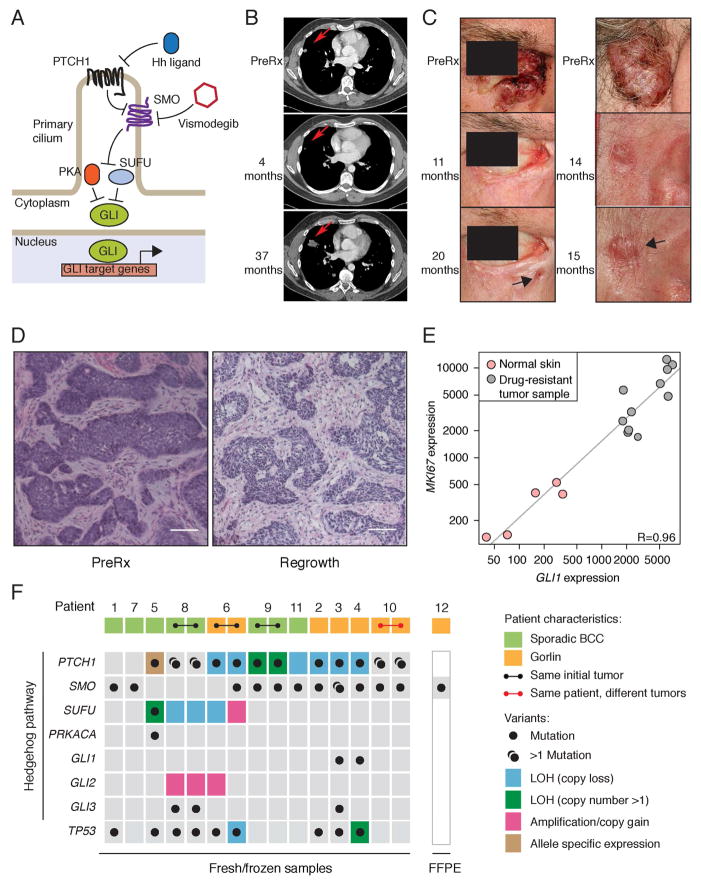
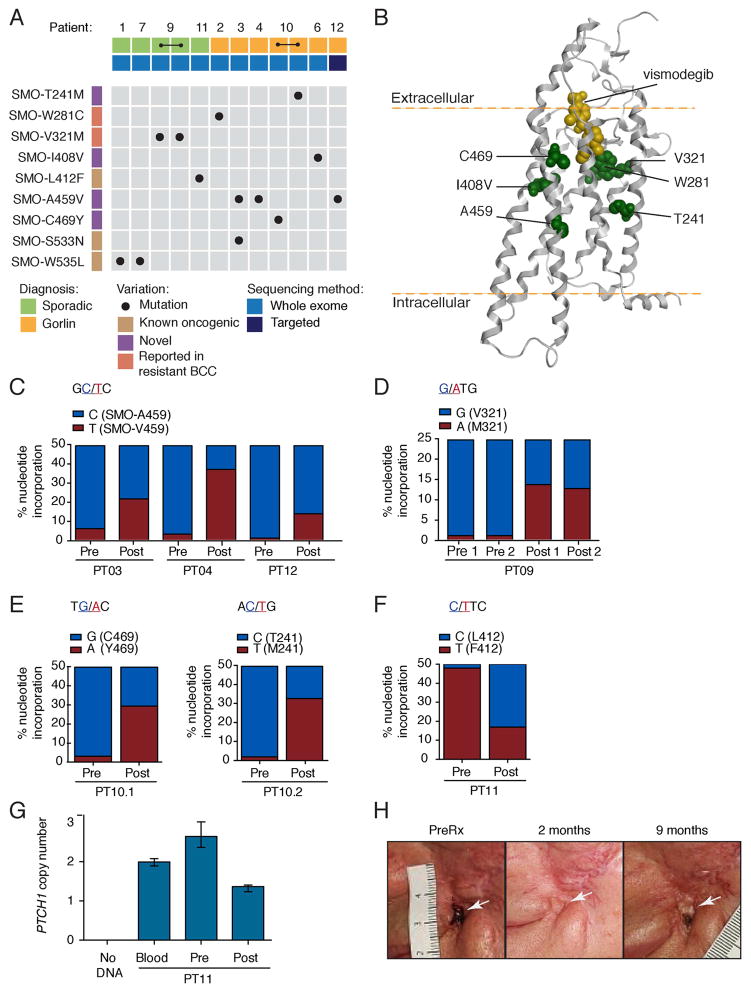
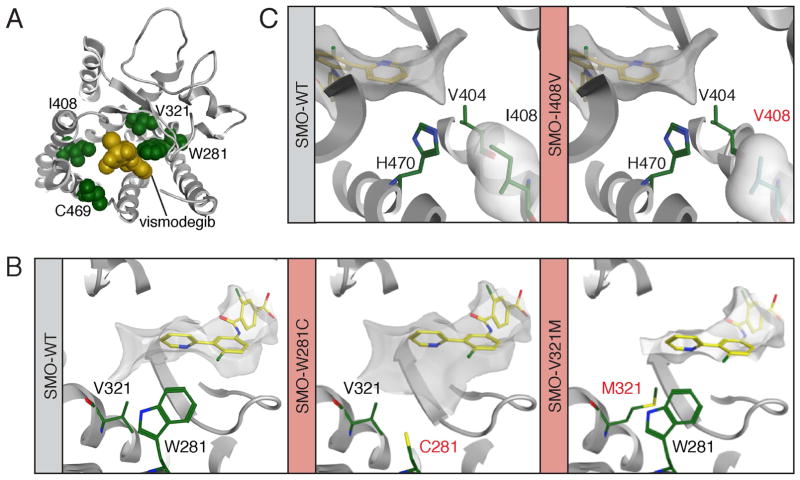
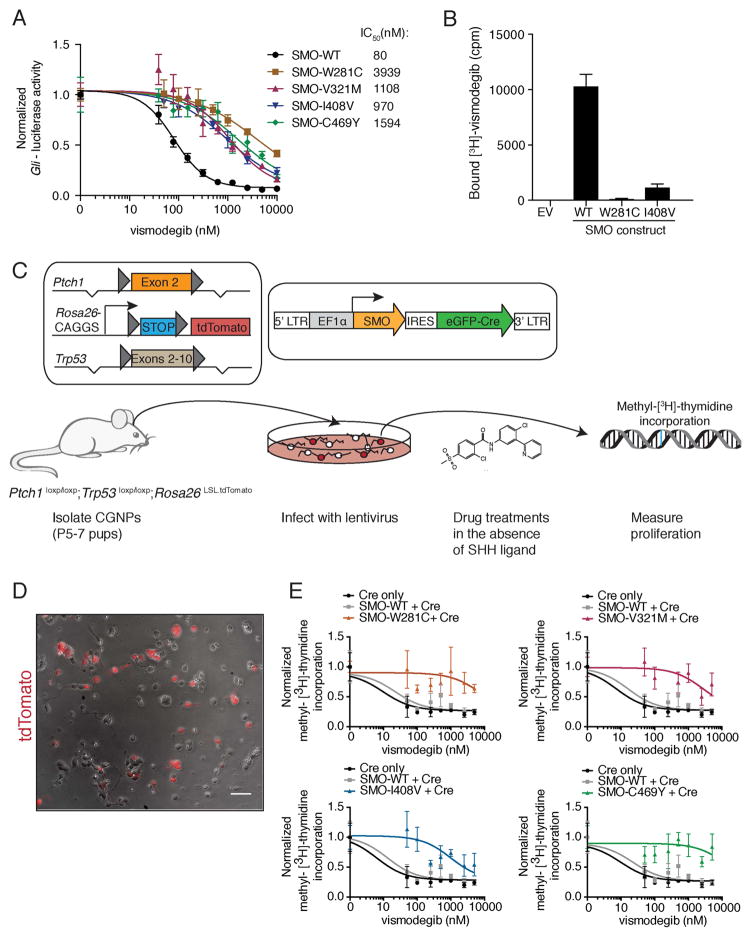
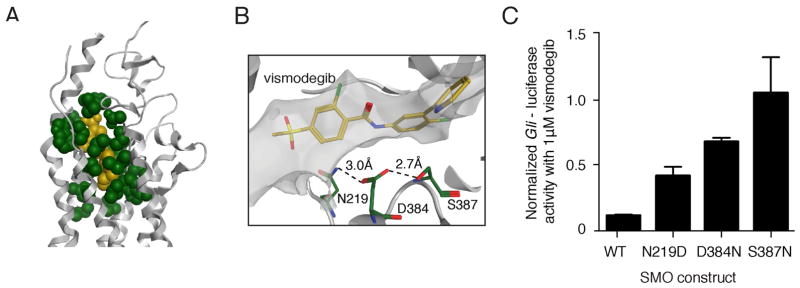
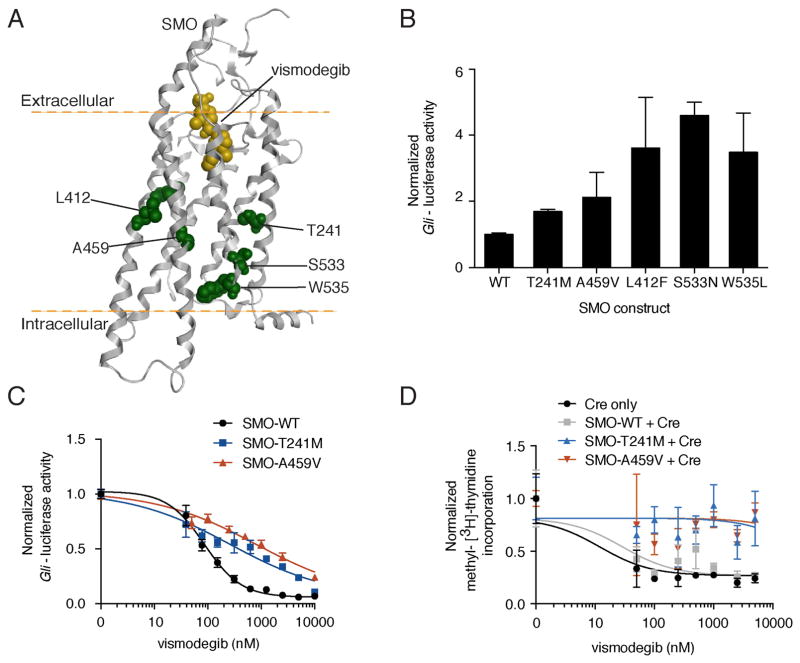
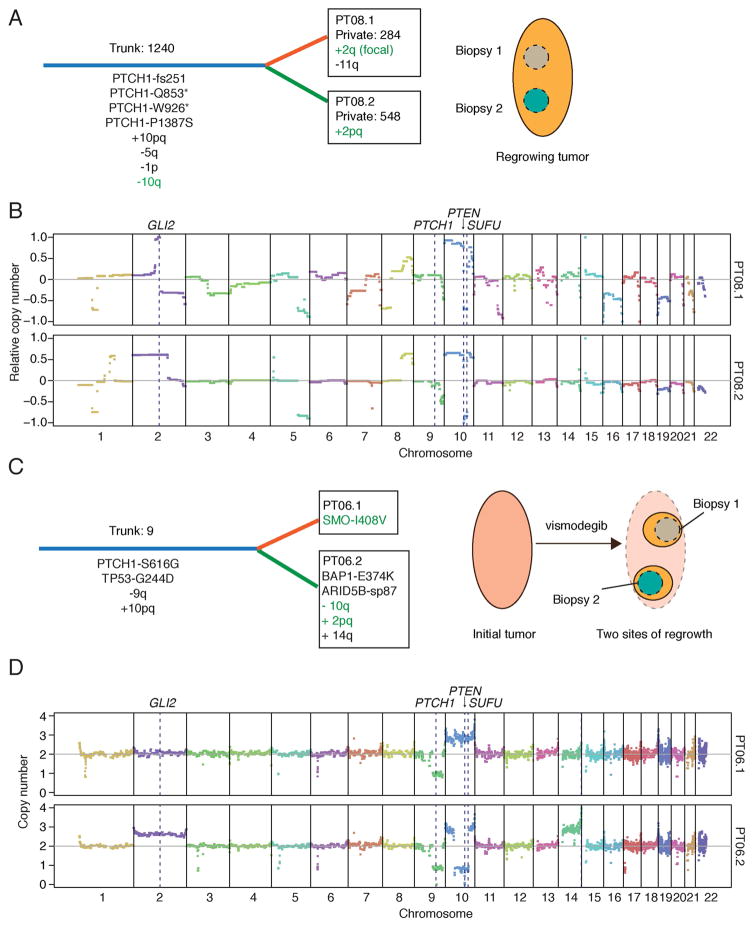
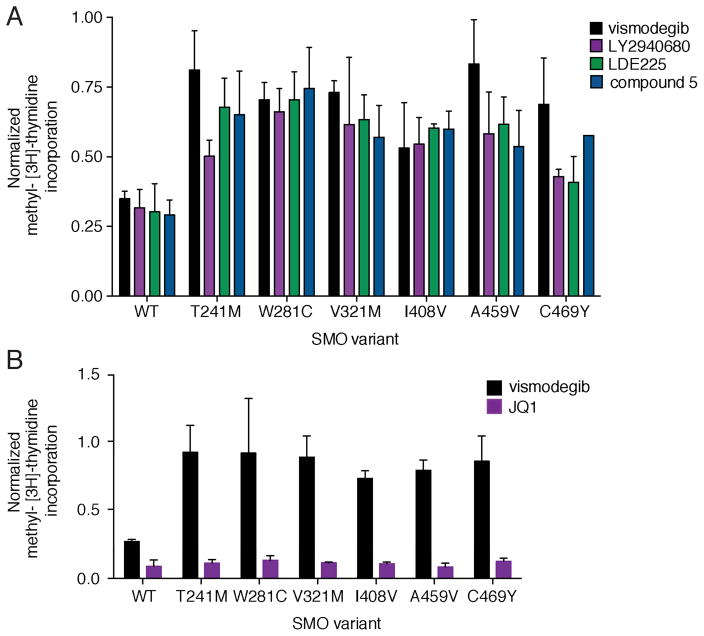
Smoothened (SMO) inhibitors are under clinical investigation for the treatment of several cancers. Vismodegib is approved for the treatment of locally advanced and metastatic basal cell carcinoma (BCC). Most BCC patients experience significant clinical benefit on vismodegib, but some develop resistance. Genomic analysis of tumor biopsies revealed that vismodegib resistance is associated with Hedgehog (Hh) pathway reactivation, predominantly through mutation of the drug target SMO and to a lesser extent through concurrent copy number changes in SUFU and GLI2. SMO mutations either directly impaired drug binding or activated SMO to varying levels. Furthermore, we found evidence for intra-tumor heterogeneity, suggesting that a combination of therapies targeting components at multiple levels of the Hh pathway is required to overcome resistance.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Vismodegib resistance in basal cell carcinoma: not a smooth fit.Cancer Cell. 2015 Mar 9;27(3):315-6. doi: 10.1016/j.ccell.2015.02.009. Cancer Cell. 2015. PMID: 25759014
-
Smoothened mutations underlie Basal cell carcinoma resistance.Cancer Discov. 2015 May;5(5):OF9. doi: 10.1158/2159-8290.CD-RW2015-053. Epub 2015 Mar 26. Cancer Discov. 2015. PMID: 25813347 No abstract available.
References
-
- Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nature medicine. 2013;19:1410–1422. - PubMed
-
- Brinkhuizen T, Reinders MG, van Geel M, Hendriksen AJ, Paulussen AD, Winnepenninckx VJ, Keymeulen KB, Soetekouw PM, van Steensel MA, Mosterd K. Acquired resistance to the Hedgehog pathway inhibitor vismodegib due to smoothened mutations in treatment of locally advanced basal cell carcinoma. Journal of the American Academy of Dermatology 2014 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
