

A comprehensive library of familial human amyotrophic lateral sclerosis induced pluripotent stem cells
- PMID: 25760436
- PMCID: PMC4356618
- DOI: 10.1371/journal.pone.0118266
A comprehensive library of familial human amyotrophic lateral sclerosis induced pluripotent stem cells
Abstract
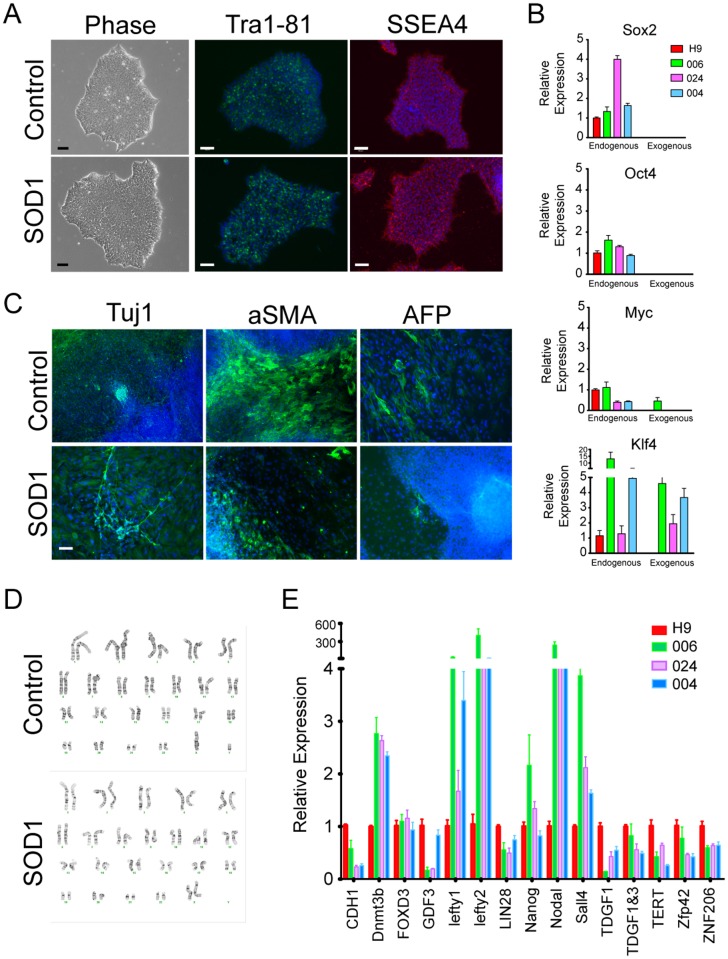
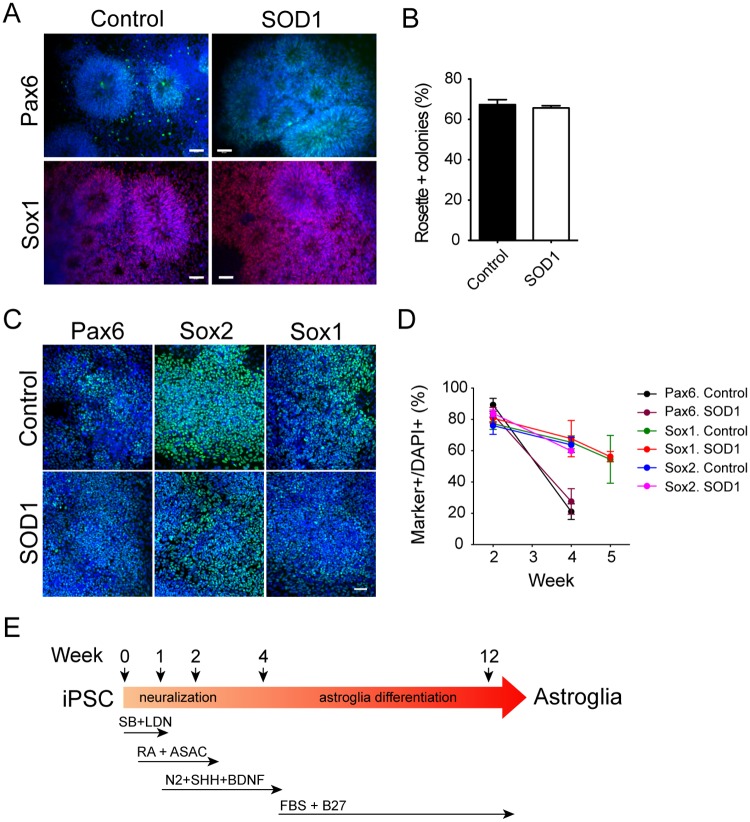
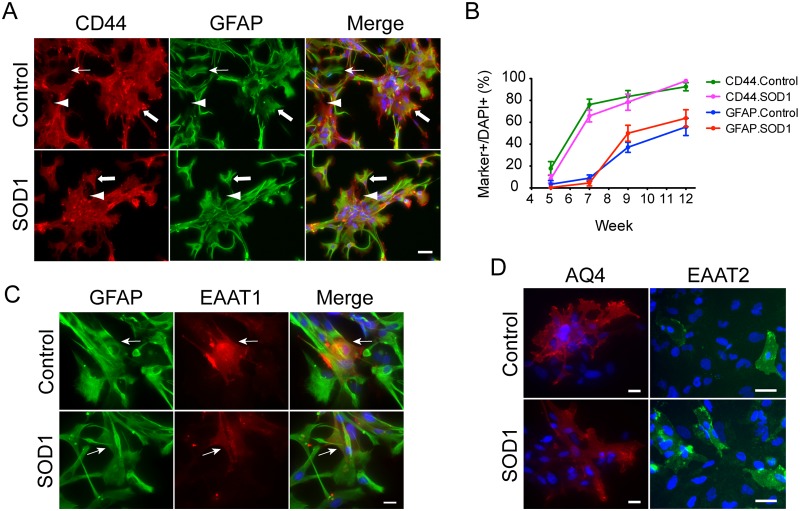
Amyotrophic lateral sclerosis is a progressive disease characterized by the loss of upper and lower motor neurons, leading to paralysis of voluntary muscles. About 10% of all ALS cases are familial (fALS), among which 15-20% are linked to Cu/Zn superoxide dismutase (SOD1) mutations, usually inherited in an autosomal dominant manner. To date only one FDA approved drug is available which increases survival moderately. Our understanding of ALS disease mechanisms is largely derived from rodent model studies, however due to the differences between rodents and humans, it is necessary to have humanized models for studies of disease pathogenesis as well as drug development. Therefore, we generated a comprehensive library of a total 22 of fALS patient-specific induced pluripotent stem cell (iPSC) lines. These cells were thoroughly characterized before being deposited into the library. The library of cells includes a variety of C9orf72 mutations, sod1 mutations, FUS, ANG and FIG4 mutations. Certain mutations are represented with more than one line, which allows for studies of variable genetic backgrounds. In addition, these iPSCs can be successfully differentiated to astroglia, a cell type known to play a critical role in ALS disease progression. This library represents a comprehensive resource that can be used for ALS disease modeling and the development of novel therapeutics.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
