

Speed of conformational change: comparing explicit and implicit solvent molecular dynamics simulations
- PMID: 25762327
- PMCID: PMC4375717
- DOI: 10.1016/j.bpj.2014.12.047
Speed of conformational change: comparing explicit and implicit solvent molecular dynamics simulations
Abstract
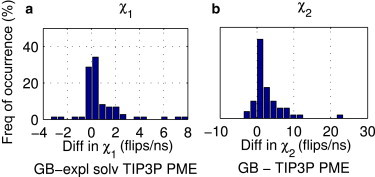
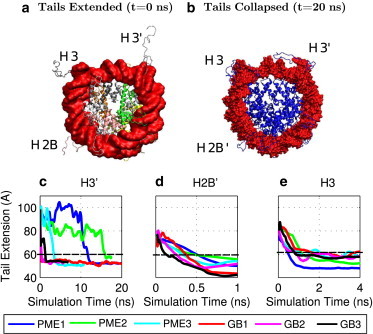
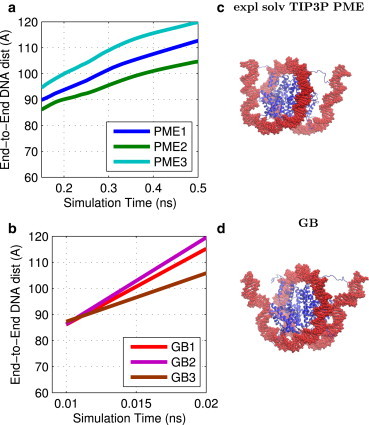
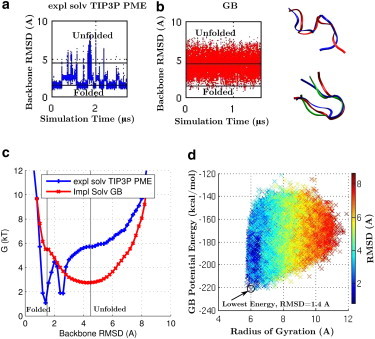
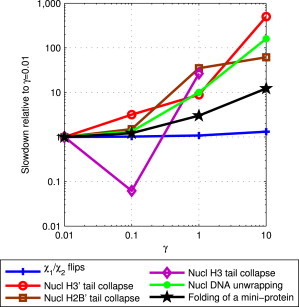
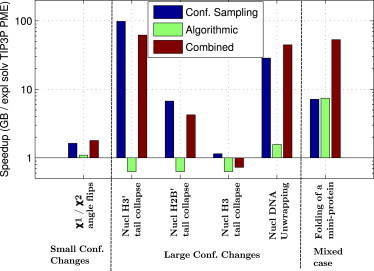
Adequate sampling of conformation space remains challenging in atomistic simulations, especially if the solvent is treated explicitly. Implicit-solvent simulations can speed up conformational sampling significantly. We compare the speed of conformational sampling between two commonly used methods of each class: the explicit-solvent particle mesh Ewald (PME) with TIP3P water model and a popular generalized Born (GB) implicit-solvent model, as implemented in the AMBER package. We systematically investigate small (dihedral angle flips in a protein), large (nucleosome tail collapse and DNA unwrapping), and mixed (folding of a miniprotein) conformational changes, with nominal simulation times ranging from nanoseconds to microseconds depending on system size. The speedups in conformational sampling for GB relative to PME simulations, are highly system- and problem-dependent. Where the simulation temperatures for PME and GB are the same, the corresponding speedups are approximately onefold (small conformational changes), between ∼1- and ∼100-fold (large changes), and approximately sevenfold (mixed case). The effects of temperature on speedup and free-energy landscapes, which may differ substantially between the solvent models, are discussed in detail for the case of miniprotein folding. In addition to speeding up conformational sampling, due to algorithmic differences, the implicit solvent model can be computationally faster for small systems or slower for large systems, depending on the number of solute and solvent atoms. For the conformational changes considered here, the combined speedups are approximately twofold, ∼1- to 60-fold, and ∼50-fold, respectively, in the low solvent viscosity regime afforded by the implicit solvent. For all the systems studied, 1) conformational sampling speedup increases as Langevin collision frequency (effective viscosity) decreases; and 2) conformational sampling speedup is mainly due to reduction in solvent viscosity rather than possible differences in free-energy landscapes between the solvent models.
Copyright © 2015 Biophysical Society. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Dror R.O., Dirks R.M., Shaw D.E. Biomolecular simulation: a computational microscope for molecular biology. Annu. Rev. Biophys. 2012;41:429–452. - PubMed
-
- Wang W., Donini O., Kollman P.A. Biomolecular simulations: recent developments in force fields, simulations of enzyme catalysis, protein-ligand, protein-protein, and protein-nucleic acid noncovalent interactions. Annu. Rev. Biophys. Biomol. Struct. 2001;30:211–243. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
