

Both mTORC1 and mTORC2 are involved in the regulation of cell adhesion
- PMID: 25762619
- PMCID: PMC4466674
- DOI: 10.18632/oncotarget.3044
Both mTORC1 and mTORC2 are involved in the regulation of cell adhesion
Abstract
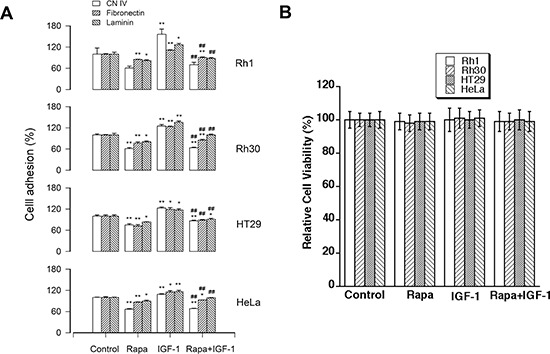
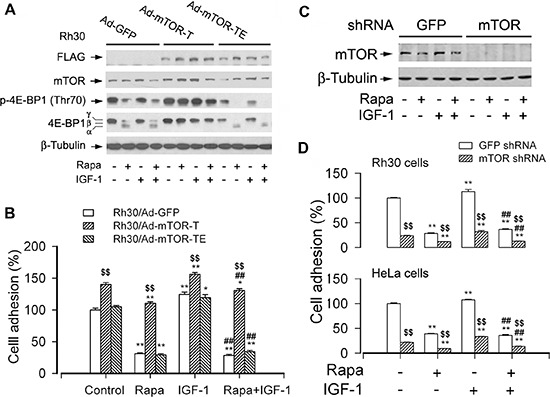
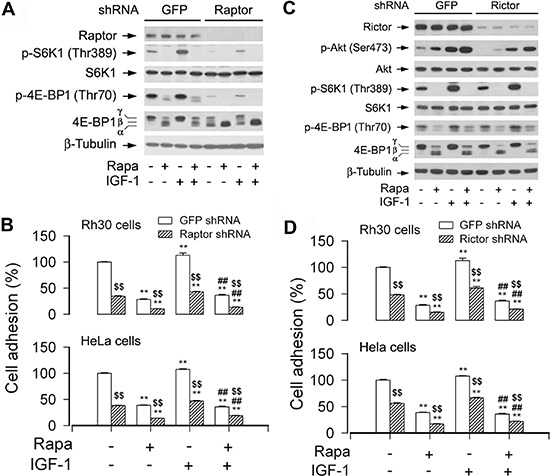
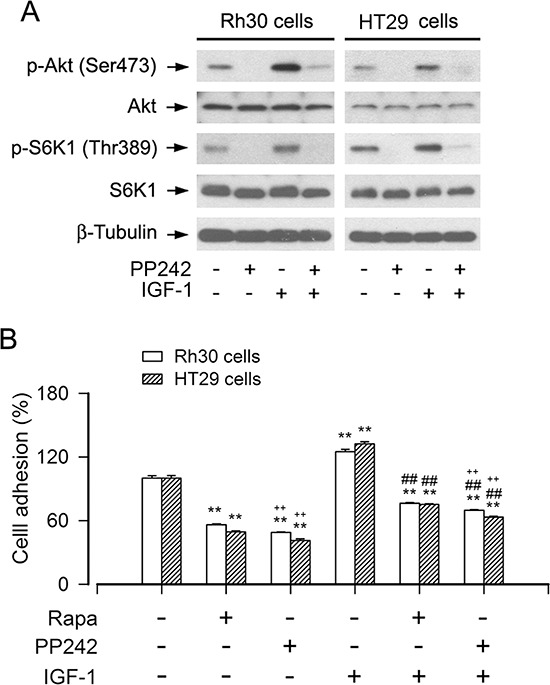
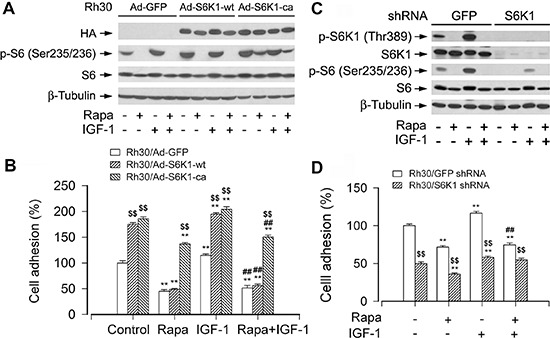
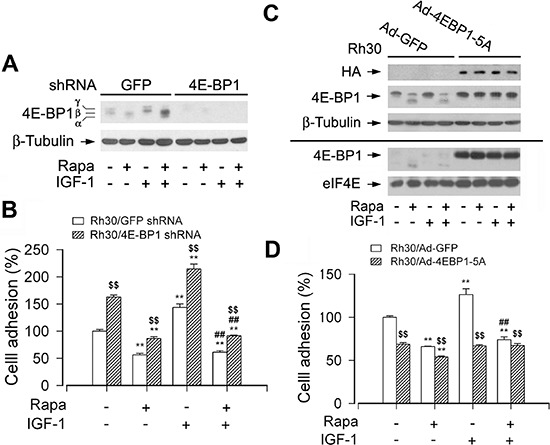
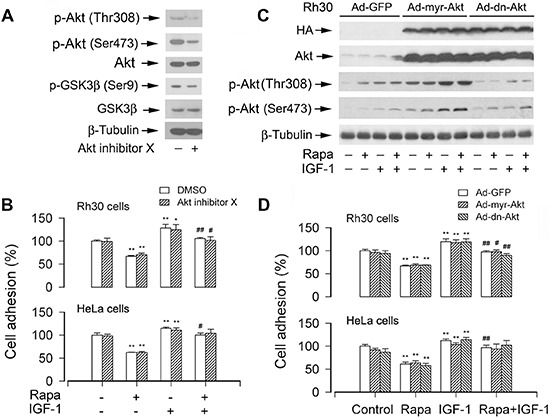
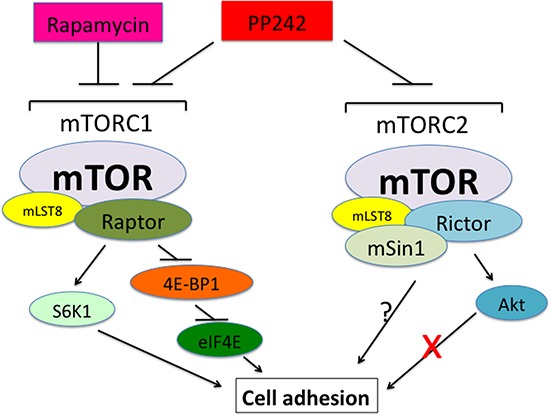
mTOR is a central controller for cell growth/proliferation and survival. Recent studies have shown that mTOR also regulates cell adhesion, yet the underlying mechanism is not known. Here we found that inhibition of mTOR by rapamycin reduced the basal or type I insulin-like growth factor (IGF-1)-stimulated adhesion of cancer cells. Further research revealed that both mTORC1 and mTORC2 were involved in the regulation of cell adhesion, as silencing expression of raptor or rictor inhibited cell adhesion. Also, PP242, an mTORC1/2 kinase inhibitor, inhibited cell adhesion more potently than rapamycin (mTORC1 inhibitor). Of interest, ectopic expression of constitutively active and rapamycin-resistant mutant of p70 kinase 1 (S6K1) or downregulation of eukaryotic initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1) conferred resistance to rapamycin inhibition of cell adhesion, whereas expression of constitutively hypophosphorylated 4E-BP1 (4EBP1-5A) or downregulation of S6K1 suppressed cell adhesion. In contrast, neither genetic manipulation of Akt activity nor pharmacological inhibition of Akt affected cell adhesion. The results suggest that both mTORC1 and mTORC2 are involved in the regulation of cell adhesion; and mTORC1 regulates cell adhesion through S6K1 and 4E-BP1 pathways, but mTORC2 regulates cell adhesion via Akt-independent mechanism.
Keywords: 4E-BP1; Akt; cell adhesion; mTOR; rapamycin.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Comment in
-
A deut of mTORC1/2 for cell adhesion.Cell Cycle. 2015;14(8):1131-2. doi: 10.1080/15384101.2015.1022075. Cell Cycle. 2015. PMID: 25789977 Free PMC article. No abstract available.
References
-
- Woodhouse EC, Chuaqui RF, Liotta LA. General mechanisms of metastasis. Cancer. 1997;80:1529–1537. - PubMed
-
- Leber MF, Efferth T. Molecular principles of cancer invasion and metastasis. Int J Oncol. 2009;34:881–895. - PubMed
-
- Cornu M, Albert V, Hall MN. mTOR in aging, metabolism, and cancer. Curr Opin Genet Dev. 2013;23:53–62. - PubMed
-
- Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–468. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
