

RNA-binding proteins in neurodegeneration: Seq and you shall receive
- PMID: 25765321
- PMCID: PMC4403644
- DOI: 10.1016/j.tins.2015.02.003
RNA-binding proteins in neurodegeneration: Seq and you shall receive
Abstract
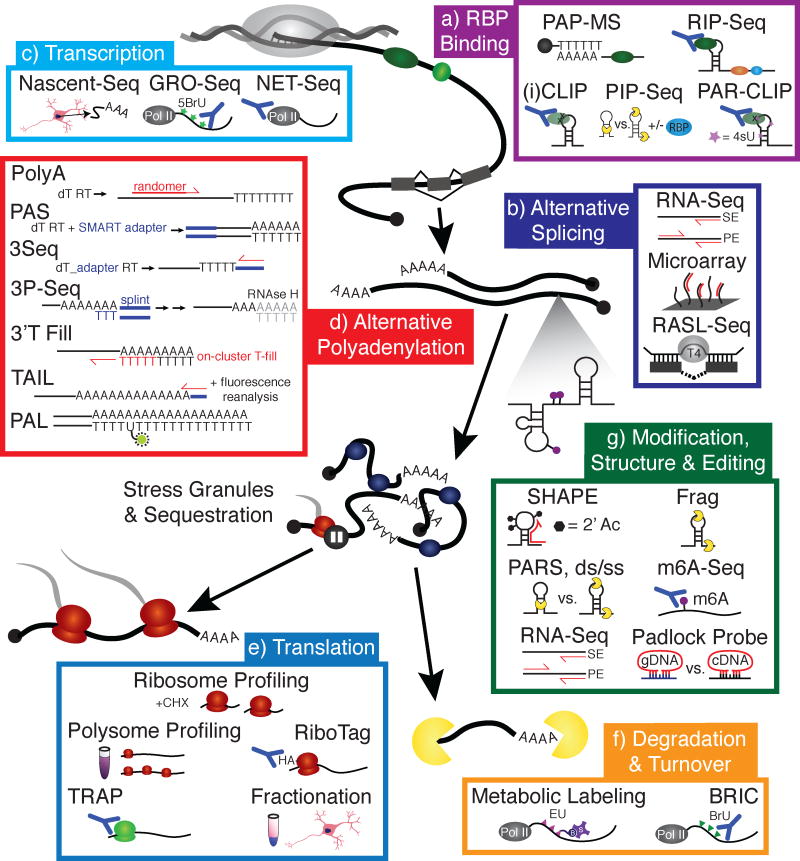
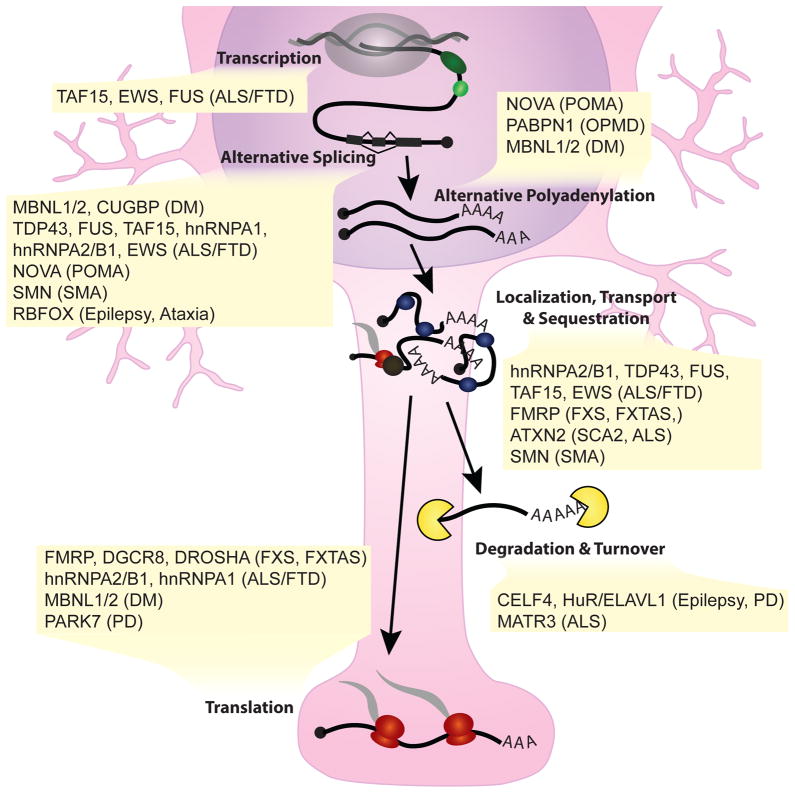
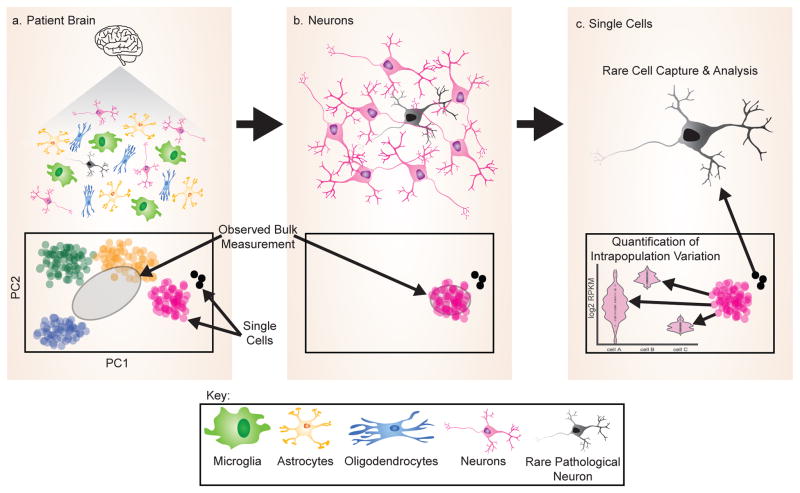
As critical players in gene regulation, RNA binding proteins (RBPs) are taking center stage in our understanding of cellular function and disease. In our era of bench-top sequencers and unprecedented computational power, biological questions can be addressed in a systematic, genome-wide manner. Development of high-throughput sequencing (Seq) methodologies provides unparalleled potential to discover new mechanisms of disease-associated perturbations of RNA homeostasis. Complementary to candidate single-gene studies, these innovative technologies may elicit the discovery of unexpected mechanisms, and enable us to determine the widespread influence of the multifunctional RBPs on their targets. Given that the disruption of RNA processing is increasingly implicated in neurological diseases, these approaches will continue to provide insights into the roles of RBPs in disease pathogenesis.
Keywords: RNA binding protein; high-throughput sequencing; neurodegeneration; polyadenylation; splicing.
Copyright © 2015 Elsevier Ltd. All rights reserved.
Figures
References
-
- Elkon R, Ugalde AP, Agami R. Alternative cleavage and polyadenylation: extent, regulation and function. Nat Rev Genet. 2013;14(7):496–506. - PubMed
-
- Balagopal V, Fluch L, Nissan T. Ways and means of eukaryotic mRNA decay. Biochim Biophys Acta. 2012;1819(6):593–603. - PubMed
-
- Frischmeyer PA, et al. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science. 2002;295(5563):2258–61. - PubMed
-
- van Hoof A, et al. Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science. 2002;295(5563):2262–4. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
