

Accurate read-based metagenome characterization using a hierarchical suite of unique signatures
- PMID: 25765641
- PMCID: PMC4446416
- DOI: 10.1093/nar/gkv180
Accurate read-based metagenome characterization using a hierarchical suite of unique signatures
Abstract
A major challenge in the field of shotgun metagenomics is the accurate identification of organisms present within a microbial community, based on classification of short sequence reads. Though existing microbial community profiling methods have attempted to rapidly classify the millions of reads output from modern sequencers, the combination of incomplete databases, similarity among otherwise divergent genomes, errors and biases in sequencing technologies, and the large volumes of sequencing data required for metagenome sequencing has led to unacceptably high false discovery rates (FDR). Here, we present the application of a novel, gene-independent and signature-based metagenomic taxonomic profiling method with significantly and consistently smaller FDR than any other available method. Our algorithm circumvents false positives using a series of non-redundant signature databases and examines Genomic Origins Through Taxonomic CHAllenge (GOTTCHA). GOTTCHA was tested and validated on 20 synthetic and mock datasets ranging in community composition and complexity, was applied successfully to data generated from spiked environmental and clinical samples, and robustly demonstrates superior performance compared with other available tools.
Published by Oxford University Press on behalf of Nucleic Acids Research 2015. This work is written by (a) US Government employee(s) and is in the public domain in the US.
Figures
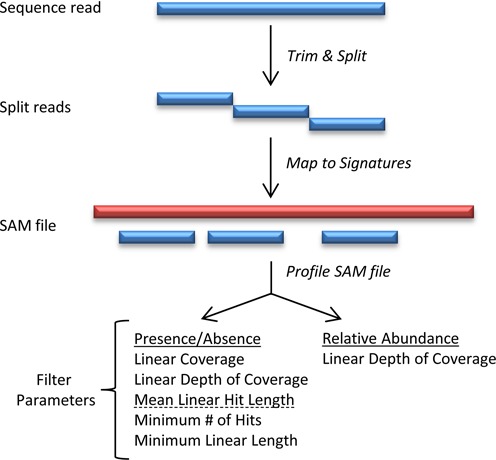
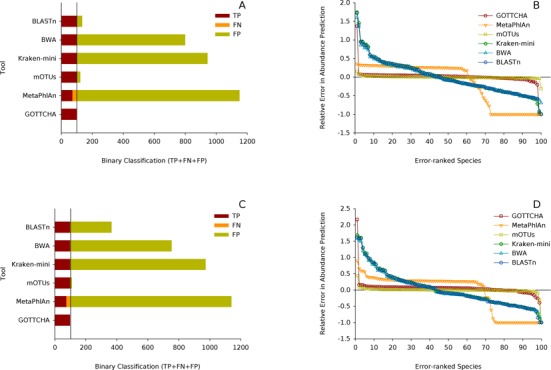
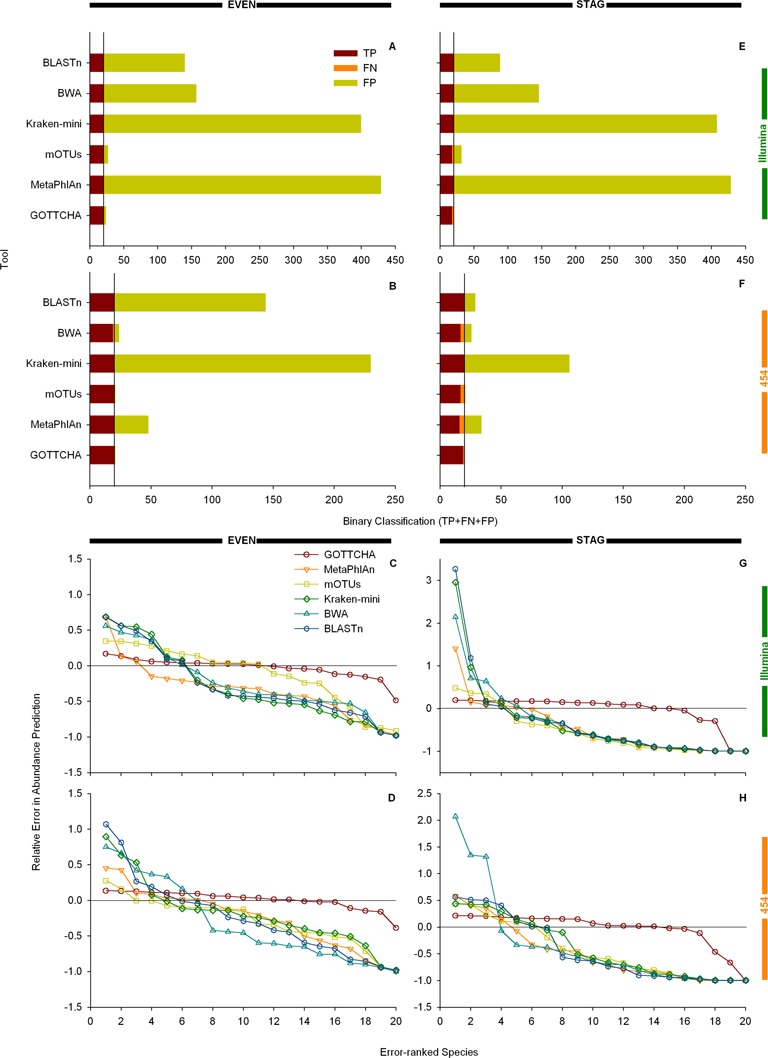
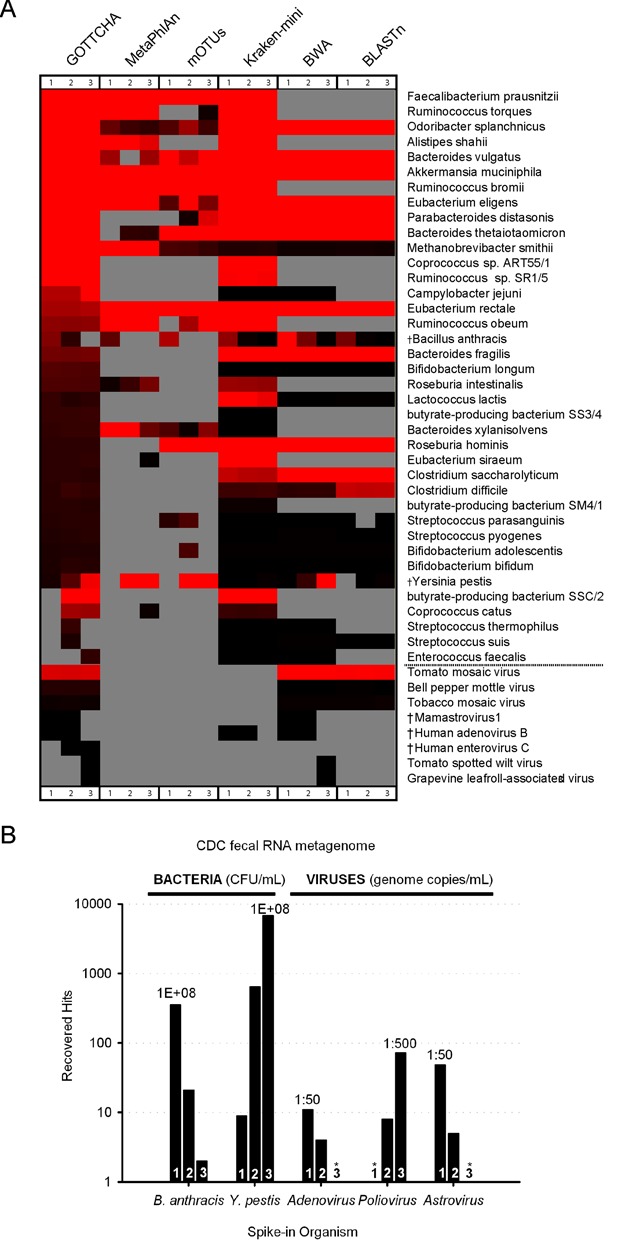
References
-
- Scholz M.B., Lo C.C., Chain P.S.G. Next generation sequencing and bioinformatic bottlenecks: the current state of metagenomic data analysis. Curr. Opin. Biotech. 2012;23:9–15. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
