

Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies
- PMID: 25766616
- PMCID: PMC4351408
- DOI: 10.1038/emm.2014.117
Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies
Abstract
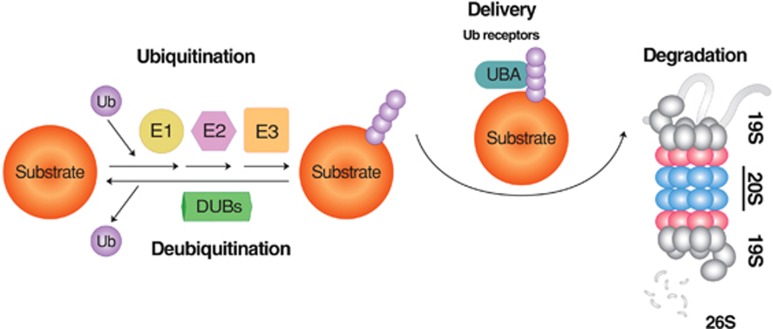
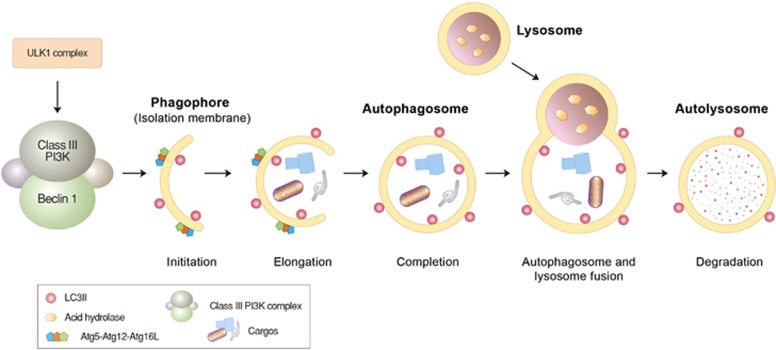
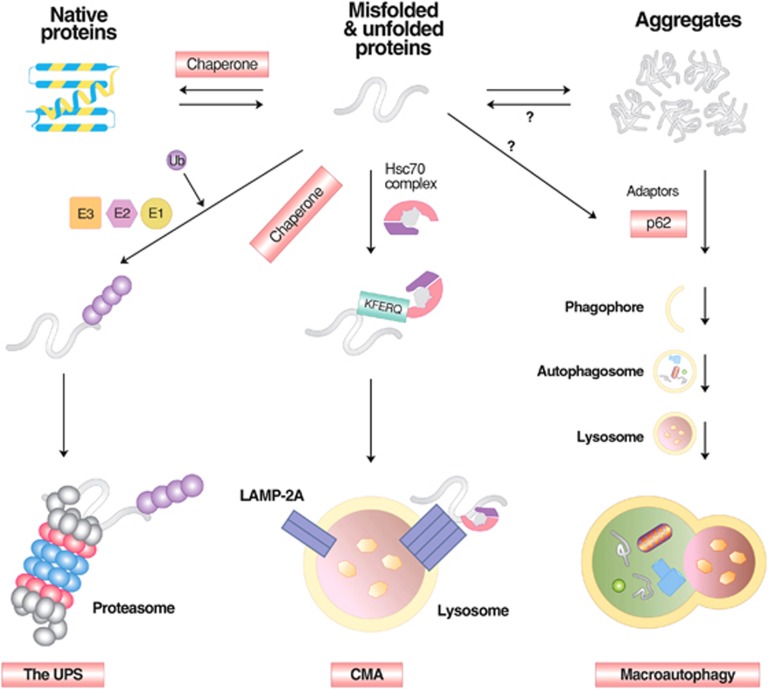
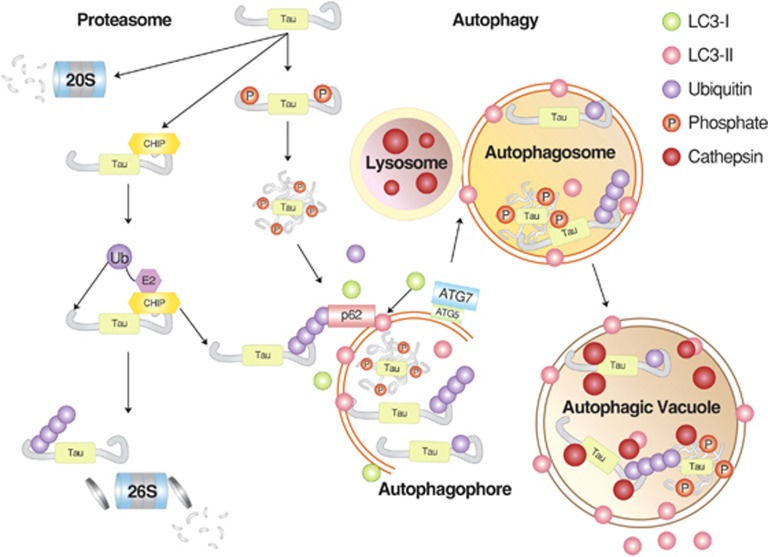
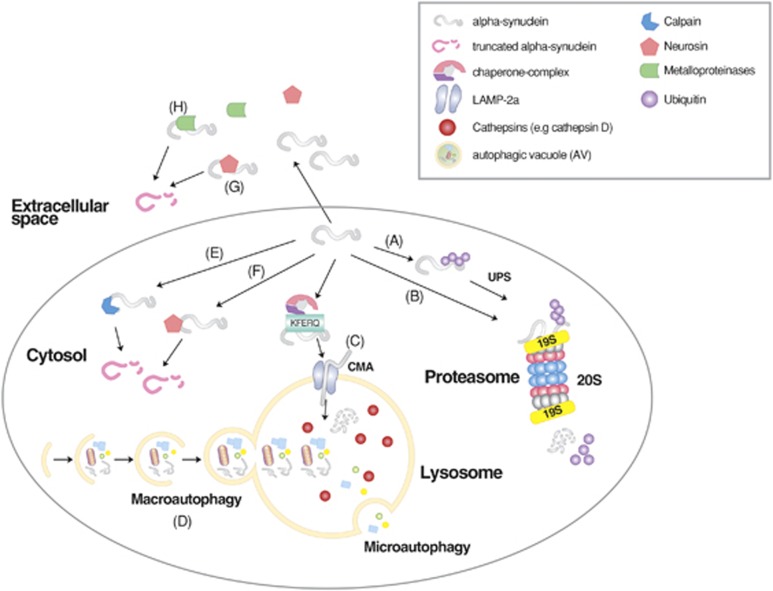
Mammalian cells remove misfolded proteins using various proteolytic systems, including the ubiquitin (Ub)-proteasome system (UPS), chaperone mediated autophagy (CMA) and macroautophagy. The majority of misfolded proteins are degraded by the UPS, in which Ub-conjugated substrates are deubiquitinated, unfolded and cleaved into small peptides when passing through the narrow chamber of the proteasome. The substrates that expose a specific degradation signal, the KFERQ sequence motif, can be delivered to and degraded in lysosomes via the CMA. Aggregation-prone substrates resistant to both the UPS and the CMA can be degraded by macroautophagy, in which cargoes are segregated into autophagosomes before degradation by lysosomal hydrolases. Although most misfolded and aggregated proteins in the human proteome can be degraded by cellular protein quality control, some native and mutant proteins prone to aggregation into β-sheet-enriched oligomers are resistant to all known proteolytic pathways and can thus grow into inclusion bodies or extracellular plaques. The accumulation of protease-resistant misfolded and aggregated proteins is a common mechanism underlying protein misfolding disorders, including neurodegenerative diseases such as Huntington's disease (HD), Alzheimer's disease (AD), Parkinson's disease (PD), prion diseases and Amyotrophic Lateral Sclerosis (ALS). In this review, we provide an overview of the proteolytic pathways in neurons, with an emphasis on the UPS, CMA and macroautophagy, and discuss the role of protein quality control in the degradation of pathogenic proteins in neurodegenerative diseases. Additionally, we examine existing putative therapeutic strategies to efficiently remove cytotoxic proteins from degenerating neurons.
Figures
References
-
- Ciechanover A. Intracellular protein degradation: from a vague idea through the lysosome and the ubiquitin–proteasome system and onto human diseases and drug targeting. Bioorg Med Chem. 2013;21:3400–3410. - PubMed
-
- Sriram SM, Kim BY, Kwon YT. The N-end rule pathway: emerging functions and molecular principles of substrate recognition. Nat Rev Mol Cell Biol. 2011;12:735–747. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
