

Enrichment of inflammatory bowel disease and colorectal cancer risk variants in colon expression quantitative trait loci
- PMID: 25766683
- PMCID: PMC4351699
- DOI: 10.1186/s12864-015-1292-z
Enrichment of inflammatory bowel disease and colorectal cancer risk variants in colon expression quantitative trait loci
Abstract
Background: Genome-wide association studies (GWAS) have identified single nucleotide polymorphisms (SNPs) associated with diseases of the colon including inflammatory bowel diseases (IBD) and colorectal cancer (CRC). However, the functional role of many of these SNPs is largely unknown and tissue-specific resources are lacking. Expression quantitative trait loci (eQTL) mapping identifies target genes of disease-associated SNPs. This study provides a comprehensive eQTL map of distal colonic samples obtained from 40 healthy African Americans and demonstrates their relevance for GWAS of colonic diseases.
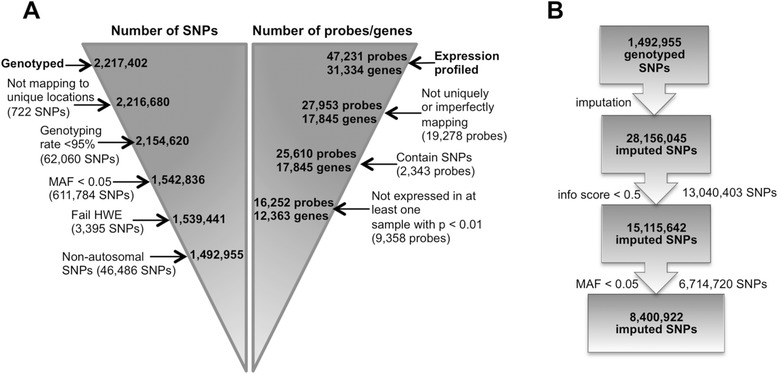
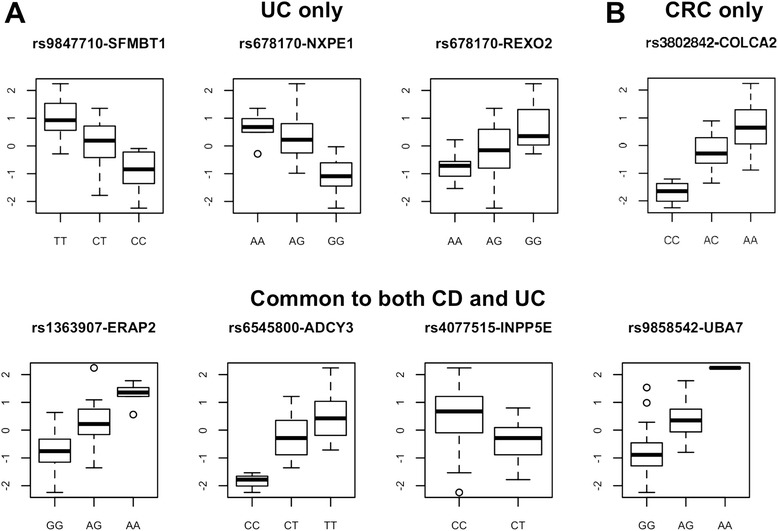
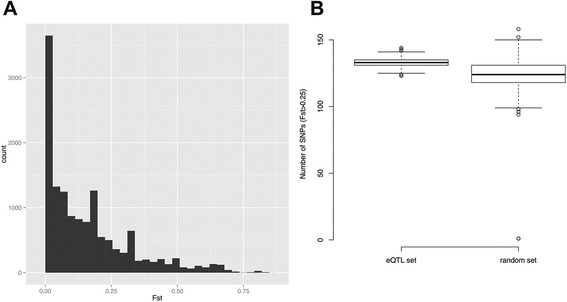
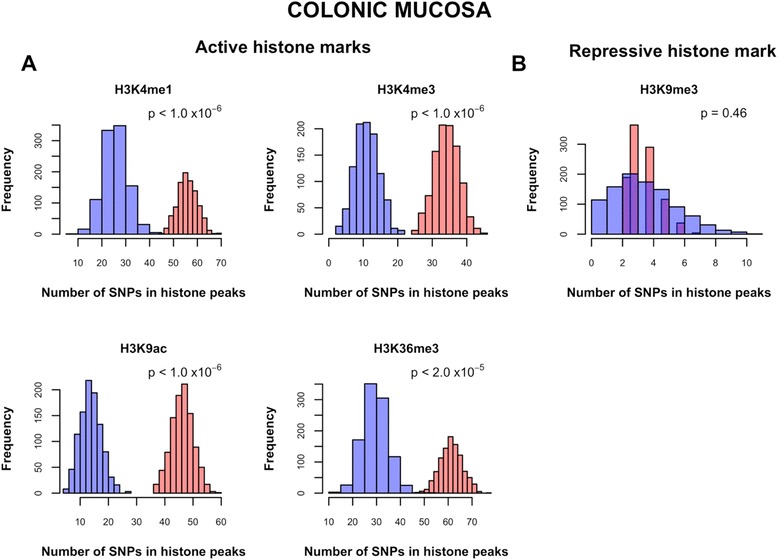
Results: 8.4 million imputed SNPs were tested for their associations with 16,252 expression probes representing 12,363 unique genes. 1,941 significant cis-eQTL, corresponding to 122 independent signals, were identified at a false discovery rate (FDR) of 0.01. Overall, among colon cis-eQTL, there was significant enrichment for GWAS variants for IBD (Crohn's disease [CD] and ulcerative colitis [UC]) and CRC as well as type 2 diabetes and body mass index. ERAP2, ADCY3, INPP5E, UBA7, SFMBT1, NXPE1 and REXO2 were identified as target genes for IBD-associated variants. The CRC-associated eQTL rs3802842 was associated with the expression of C11orf93 (COLCA2). Enrichment of colon eQTL near transcription start sites and for active histone marks was demonstrated, and eQTL with high population differentiation were identified.
Conclusions: Through the comprehensive study of eQTL in the human colon, this study identified novel target genes for IBD- and CRC-associated genetic variants. Moreover, bioinformatic characterization of colon eQTL provides a tissue-specific tool to improve understanding of biological differences in diseases between different ethnic groups.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
