

Inflammation, immunity, and hypertensive end-organ damage
- PMID: 25767287
- PMCID: PMC4535695
- DOI: 10.1161/CIRCRESAHA.116.303697
Inflammation, immunity, and hypertensive end-organ damage
Abstract
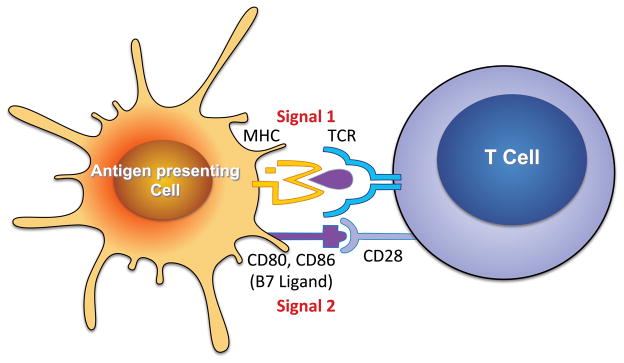
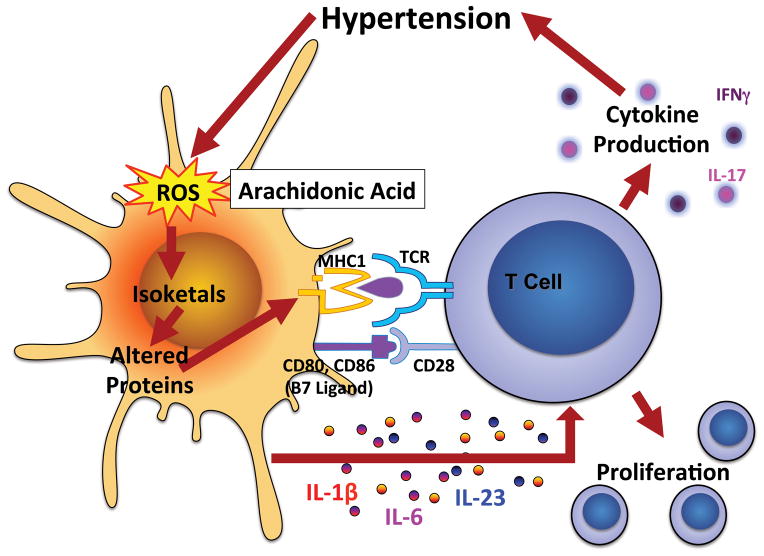
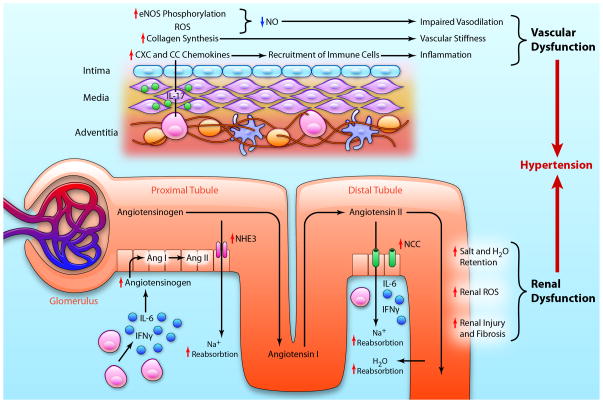
For >50 years, it has been recognized that immunity contributes to hypertension. Recent data have defined an important role of T cells and various T cell-derived cytokines in several models of experimental hypertension. These studies have shown that stimuli like angiotensin II, deoxycorticosterone acetate-salt, and excessive catecholamines lead to formation of effector like T cells that infiltrate the kidney and perivascular regions of both large arteries and arterioles. There is also accumulation of monocyte/macrophages in these regions. Cytokines released from these cells, including interleukin-17, interferon-γ, tumor necrosis factorα, and interleukin-6 promote both renal and vascular dysfunction and damage, leading to enhanced sodium retention and increased systemic vascular resistance. The renal effects of these cytokines remain to be fully defined, but include enhanced formation of angiotensinogen, increased sodium reabsorption, and increased renal fibrosis. Recent experiments have defined a link between oxidative stress and immune activation in hypertension. These have shown that hypertension is associated with formation of reactive oxygen species in dendritic cells that lead to formation of gamma ketoaldehydes, or isoketals. These rapidly adduct to protein lysines and are presented by dendritic cells as neoantigens that activate T cells and promote hypertension. Thus, cells of both the innate and adaptive immune system contribute to end-organ damage and dysfunction in hypertension. Therapeutic interventions to reduce activation of these cells may prove beneficial in reducing end-organ damage and preventing consequences of hypertension, including myocardial infarction, heart failure, renal failure, and stroke.
Keywords: angiotensin II; antigen presenting cell; cytokines; effector T cell; nitric oxide synthase; sodium.
© 2015 American Heart Association, Inc.
Conflict of interest statement
Figures
References
-
- White FN, Grollman A. Autoimmune Factors Associated with Infarction of the Kidney. Nephron. 1964;204:93–102. - PubMed
-
- Okuda T, Grollman A. Passive transfer of autoimmune induced hypertension in the rat by lymph node cells. Tex Rep Biol Med. 1967;25:257–64. - PubMed
-
- Olsen F. Type and course of the inflammatory cellular reaction in acute angiotensin-hypertensive vascular disease in rats. Acta Pathol Microbiol Scand A. 1970;78:143–50. - PubMed
-
- Olsen F. Inflammatory cellular reaction in hypertensive vascular disease in man. Acta Pathol Microbiol Scand A. 1972;80:253–6. - PubMed
-
- Takeichi N, Hamada J, Takimoto M, Fujiwara K, Kobayashi H. Depression of T cell-mediated immunity and enhancement of autoantibody production by natural infection with microorganisms in spontaneously hypertensive rats (SHR) Microbiol Immunol. 1988;32:1235–44. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- K08HL121671/HL/NHLBI NIH HHS/United States
- R01 HL039006/HL/NHLBI NIH HHS/United States
- P01HL095070/HL/NHLBI NIH HHS/United States
- R01 HL108701/HL/NHLBI NIH HHS/United States
- K08 HL121671/HL/NHLBI NIH HHS/United States
- P01 HL095070/HL/NHLBI NIH HHS/United States
- P01 HL058000/HL/NHLBI NIH HHS/United States
- R01HL108701/HL/NHLBI NIH HHS/United States
- P01 GM015431/GM/NIGMS NIH HHS/United States
- T32 HL069765/HL/NHLBI NIH HHS/United States
- R01 HL125865/HL/NHLBI NIH HHS/United States
- R01 HL105294/HL/NHLBI NIH HHS/United States
- R01HL039006/HL/NHLBI NIH HHS/United States
- P01GM015431/GM/NIGMS NIH HHS/United States
- T32 HL69765/HL/NHLBI NIH HHS/United States
- R01HL105294/HL/NHLBI NIH HHS/United States
- K01 HL130497/HL/NHLBI NIH HHS/United States
- P01HL058000/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
