

Molecular mechanisms underlying β-adrenergic receptor-mediated cross-talk between sympathetic neurons and immune cells
- PMID: 25768345
- PMCID: PMC4394497
- DOI: 10.3390/ijms16035635
Molecular mechanisms underlying β-adrenergic receptor-mediated cross-talk between sympathetic neurons and immune cells
Abstract
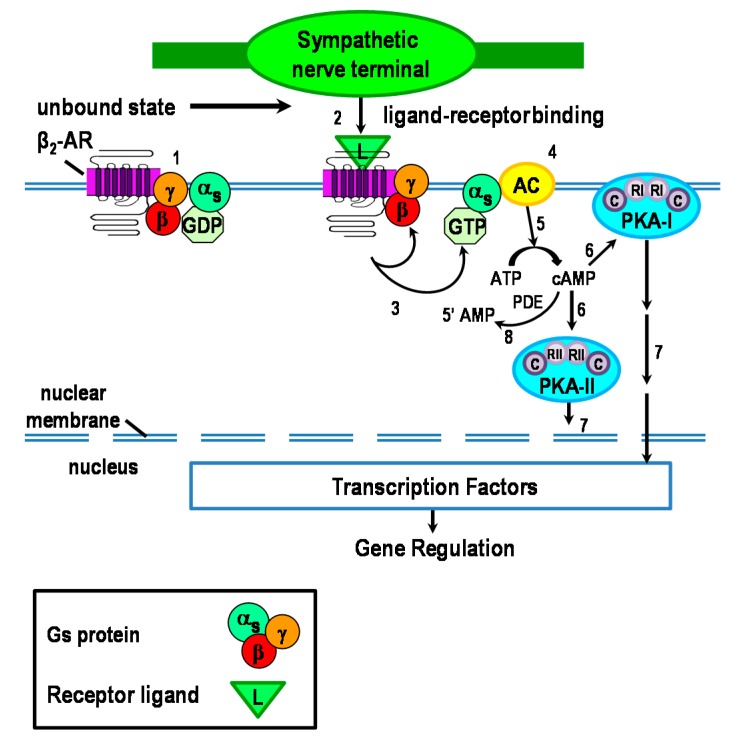
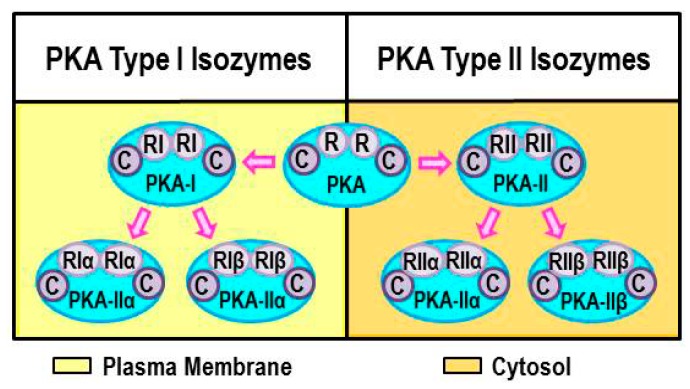
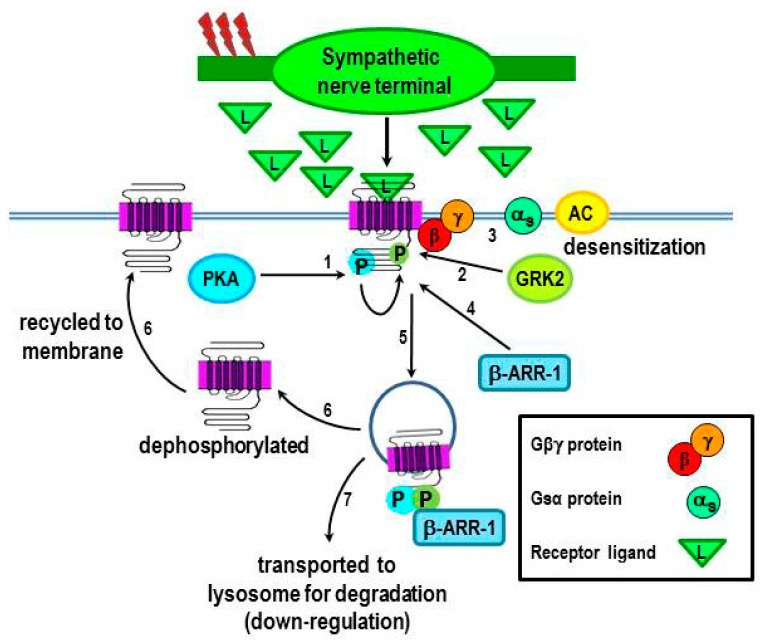
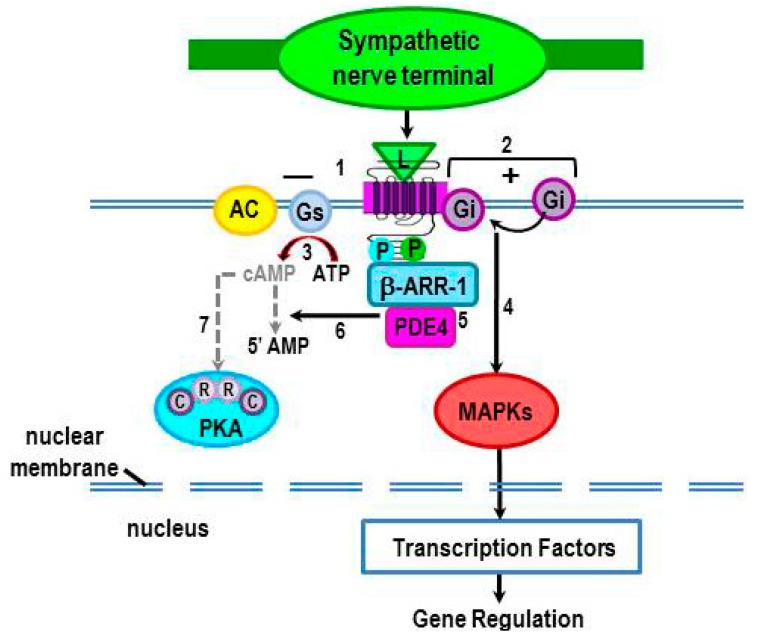
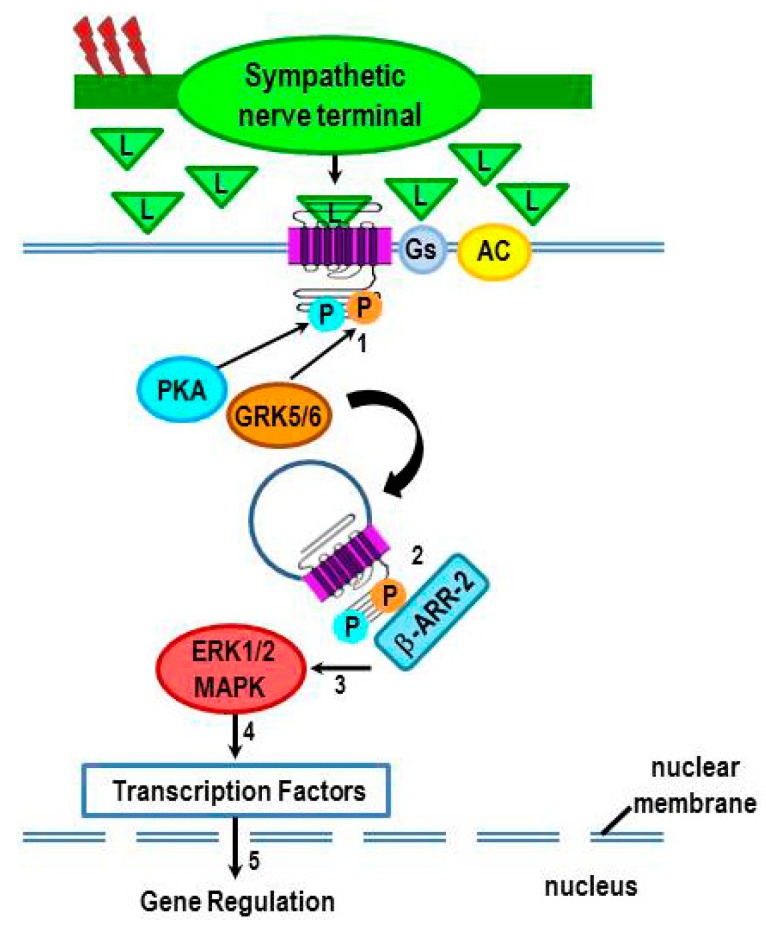
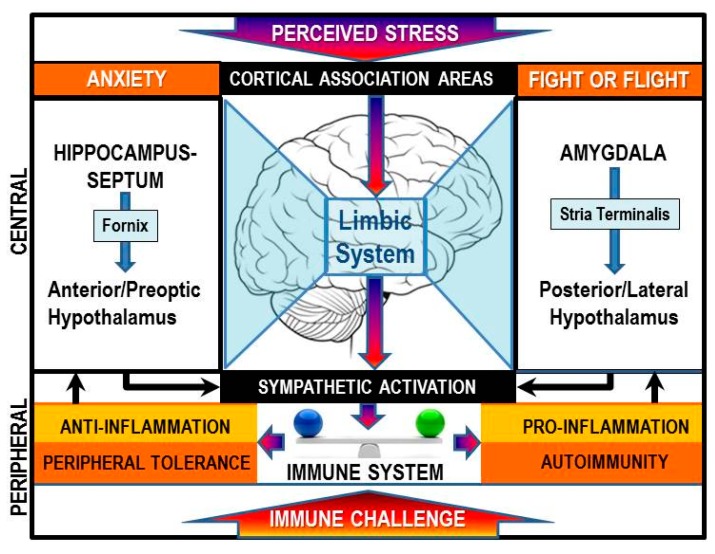
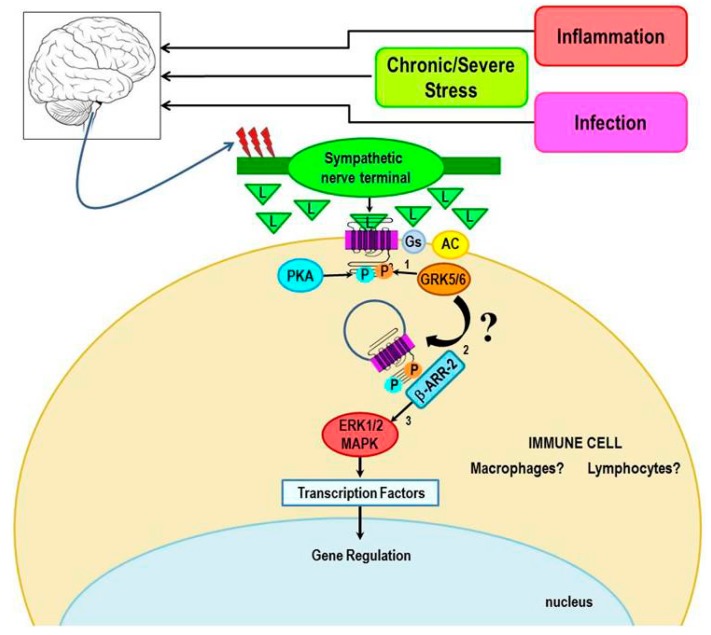
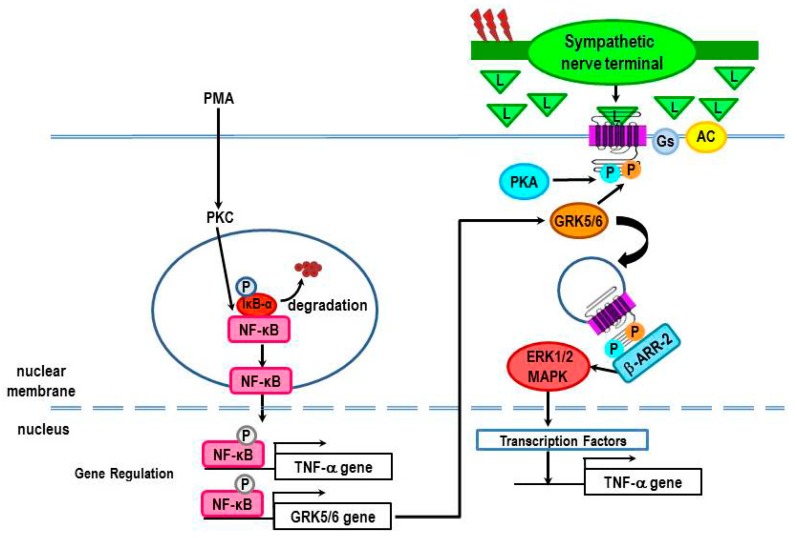
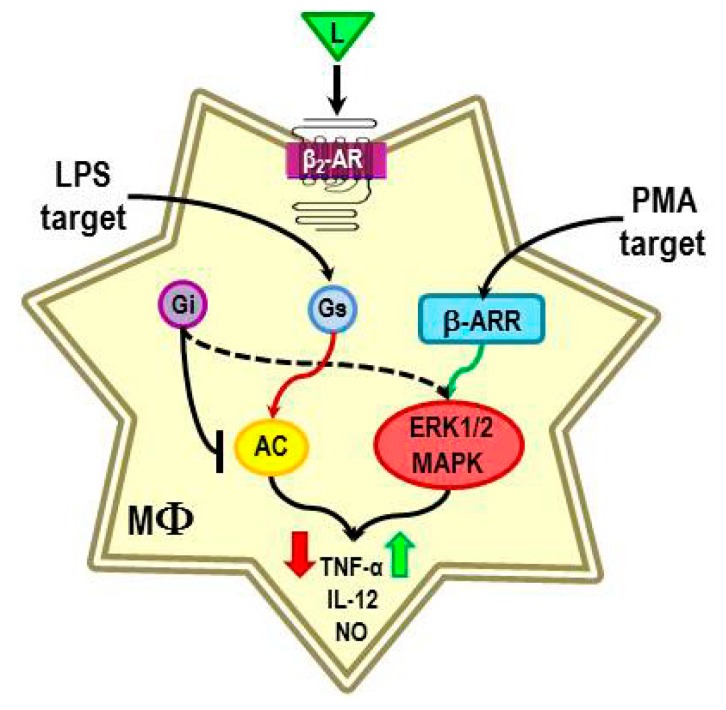
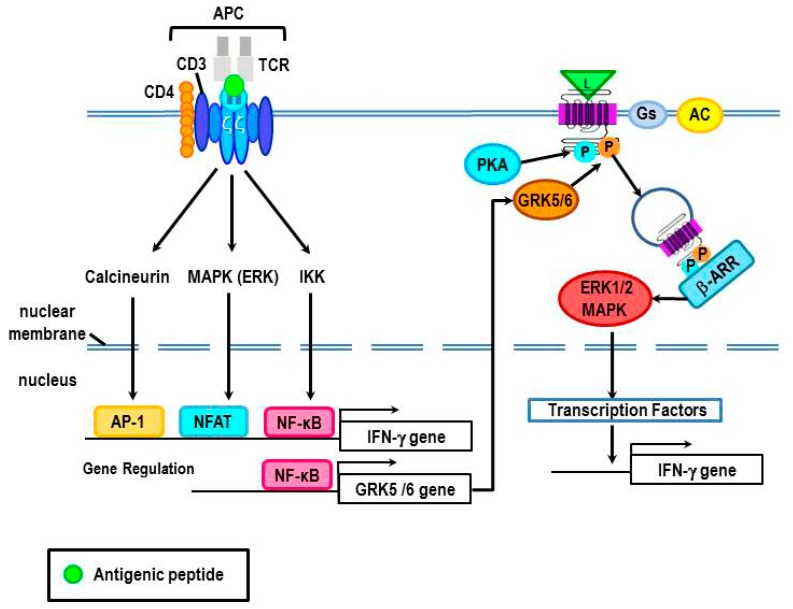
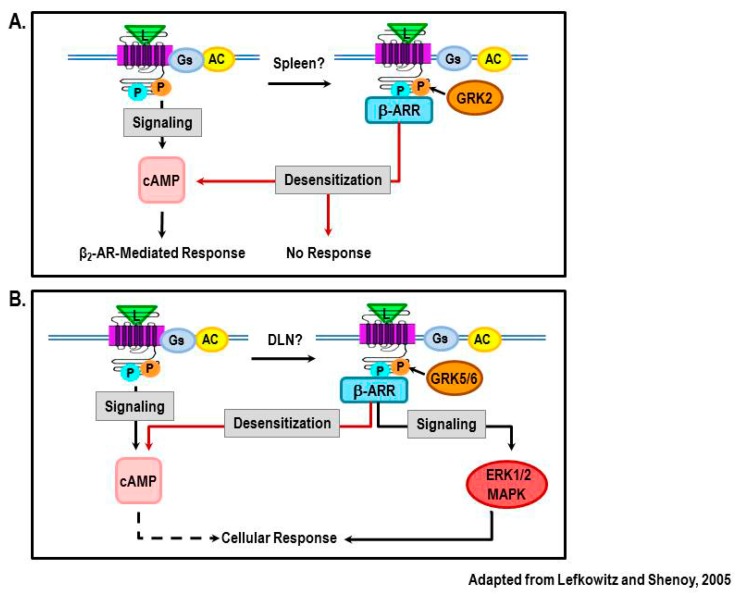
Cross-talk between the sympathetic nervous system (SNS) and immune system is vital for health and well-being. Infection, tissue injury and inflammation raise firing rates of sympathetic nerves, increasing their release of norepinephrine (NE) in lymphoid organs and tissues. NE stimulation of β2-adrenergic receptors (ARs) in immune cells activates the cAMP-protein kinase A (PKA) intracellular signaling pathway, a pathway that interfaces with other signaling pathways that regulate proliferation, differentiation, maturation and effector functions in immune cells. Immune-SNS cross-talk is required to maintain homeostasis under normal conditions, to develop an immune response of appropriate magnitude after injury or immune challenge, and subsequently restore homeostasis. Typically, β2-AR-induced cAMP is immunosuppressive. However, many studies report actions of β2-AR stimulation in immune cells that are inconsistent with typical cAMP-PKA signal transduction. Research during the last decade in non-immune organs, has unveiled novel alternative signaling mechanisms induced by β2-AR activation, such as a signaling switch from cAMP-PKA to mitogen-activated protein kinase (MAPK) pathways. If alternative signaling occurs in immune cells, it may explain inconsistent findings of sympathetic regulation of immune function. Here, we review β2-AR signaling, assess the available evidence for alternative signaling in immune cells, and provide insight into the circumstances necessary for "signal switching" in immune cells.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
