

Comparative analysis of field-isolate and monkey-adapted Plasmodium vivax genomes
- PMID: 25768941
- PMCID: PMC4358935
- DOI: 10.1371/journal.pntd.0003566
Comparative analysis of field-isolate and monkey-adapted Plasmodium vivax genomes
Abstract
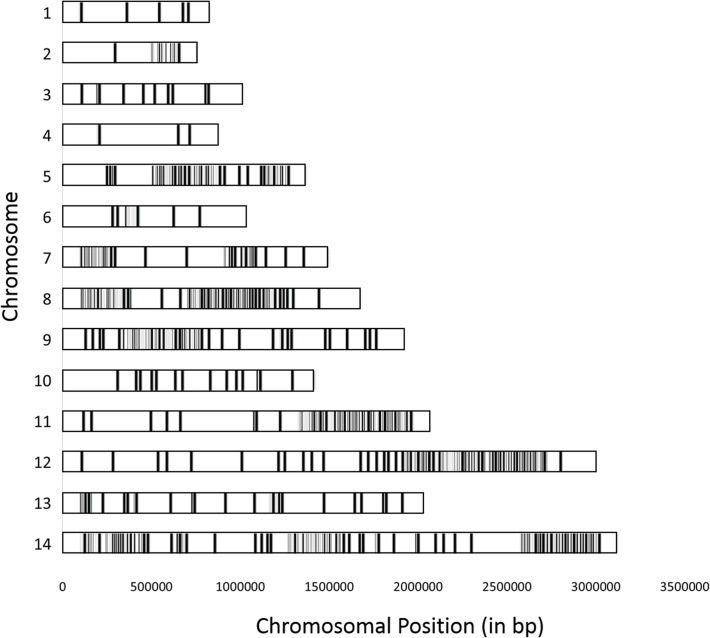
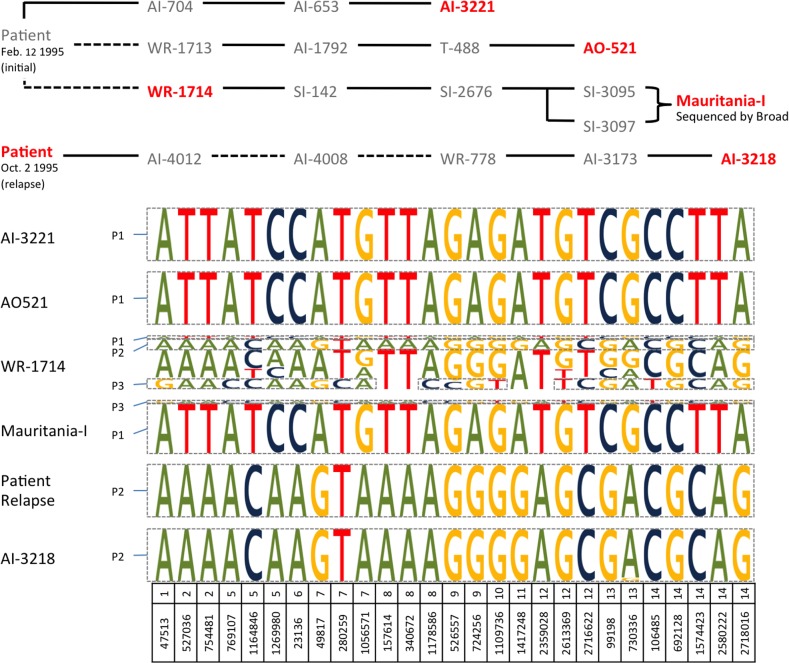
Significant insights into the biology of Plasmodium vivax have been gained from the ability to successfully adapt human infections to non-human primates. P. vivax strains grown in monkeys serve as a renewable source of parasites for in vitro and ex vivo experimental studies and functional assays, or for studying in vivo the relapse characteristics, mosquito species compatibilities, drug susceptibility profiles or immune responses towards potential vaccine candidates. Despite the importance of these studies, little is known as to how adaptation to a different host species may influence the genome of P. vivax. In addition, it is unclear whether these monkey-adapted strains consist of a single clonal population of parasites or if they retain the multiclonal complexity commonly observed in field isolates. Here we compare the genome sequences of seven P. vivax strains adapted to New World monkeys with those of six human clinical isolates collected directly in the field. We show that the adaptation of P. vivax parasites to monkey hosts, and their subsequent propagation, did not result in significant modifications of their genome sequence and that these monkey-adapted strains recapitulate the genomic diversity of field isolates. Our analyses also reveal that these strains are not always genetically homogeneous and should be analyzed cautiously. Overall, our study provides a framework to better leverage this important research material and fully utilize this resource for improving our understanding of P. vivax biology.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
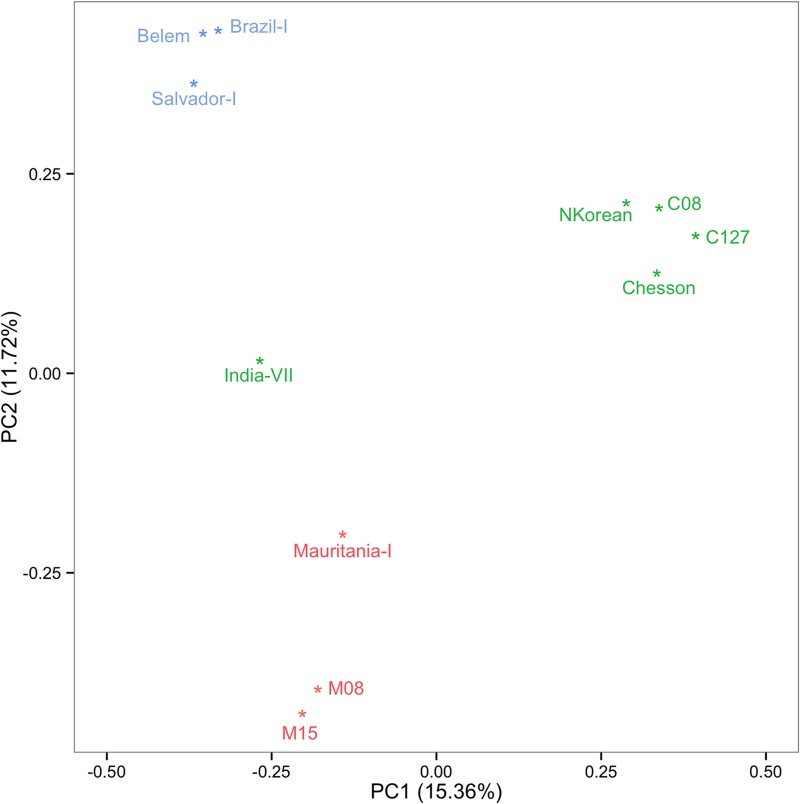
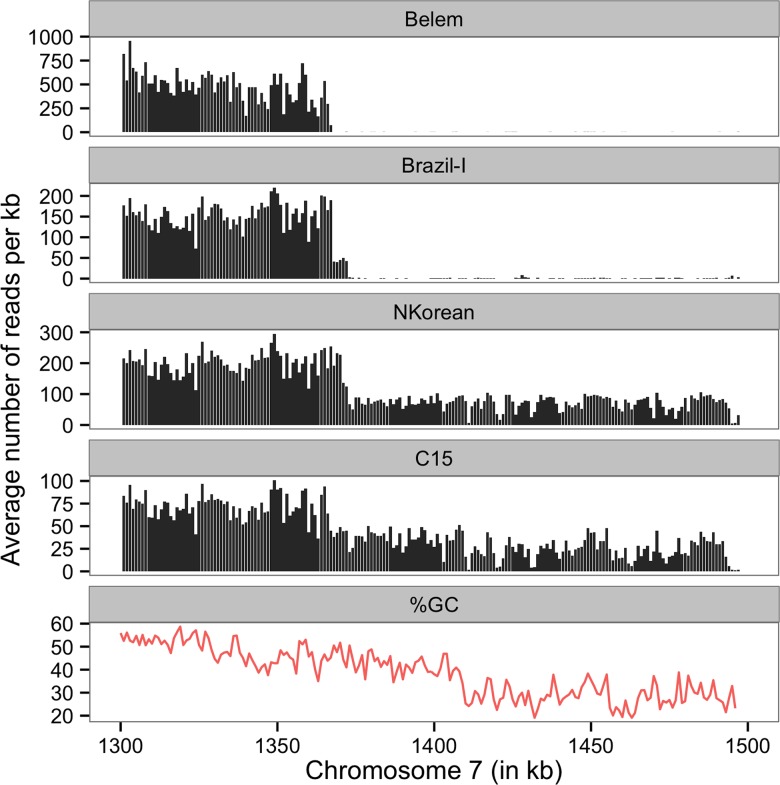
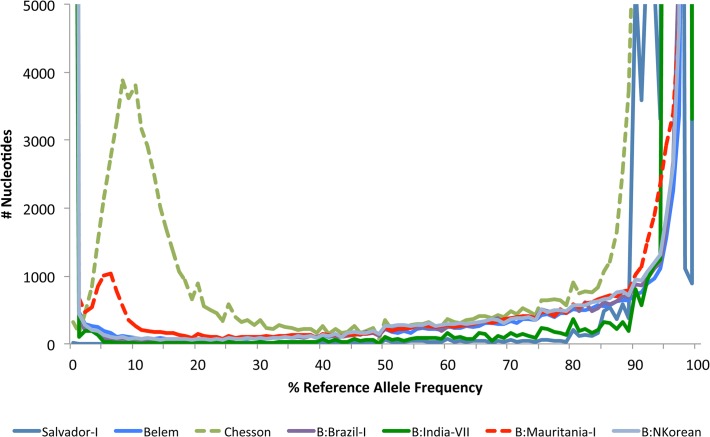
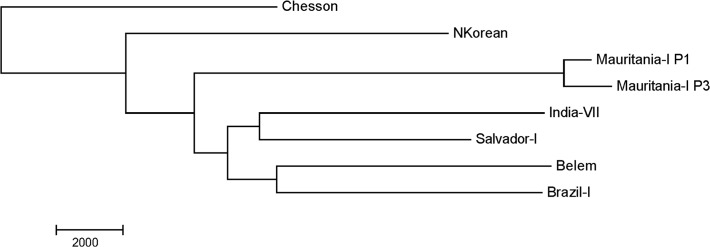
References
-
- Dharia NV, Bright aT, Westenberger SJ, Barnes SW, Batalov S, et al. (2010) Whole-genome sequencing and microarray analysis of ex vivo Plasmodium vivax reveal selective pressure on putative drug resistance genes. Proceedings of the National Academy of Sciences of the United States of America 107: 20045–20050. 10.1073/pnas.1003776107 - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
