

Autoimmune host-microbiota interactions at barrier sites and beyond
- PMID: 25771098
- PMCID: PMC5918312
- DOI: 10.1016/j.molmed.2015.02.006
Autoimmune host-microbiota interactions at barrier sites and beyond
Abstract
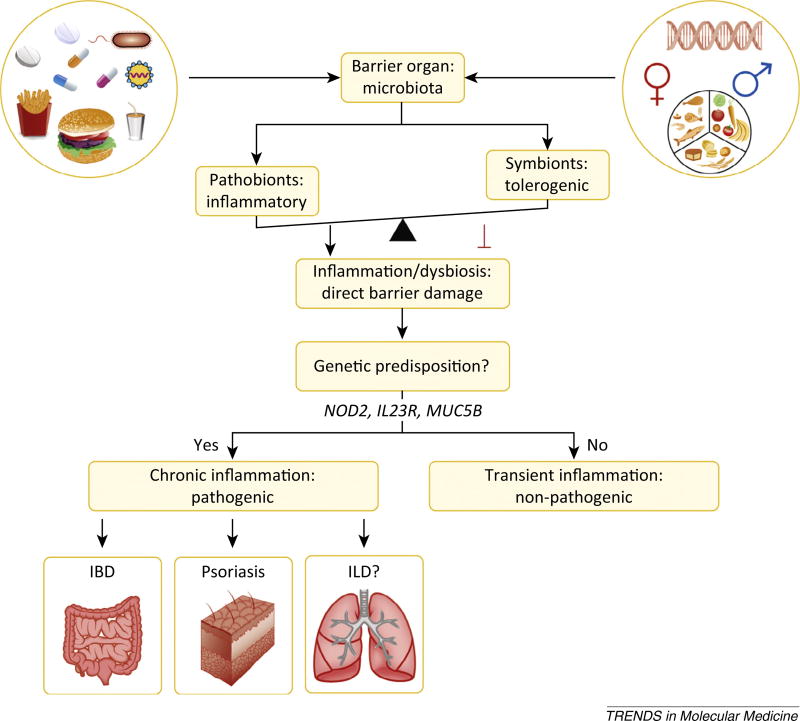
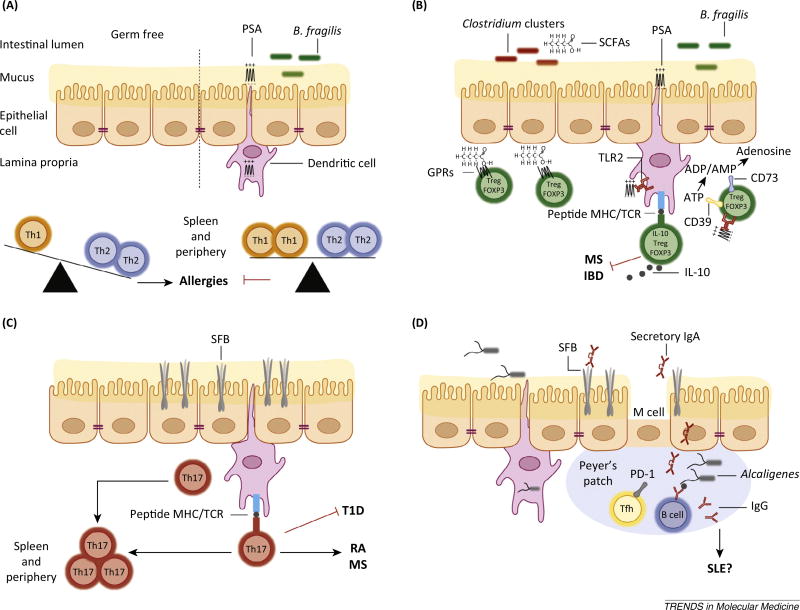
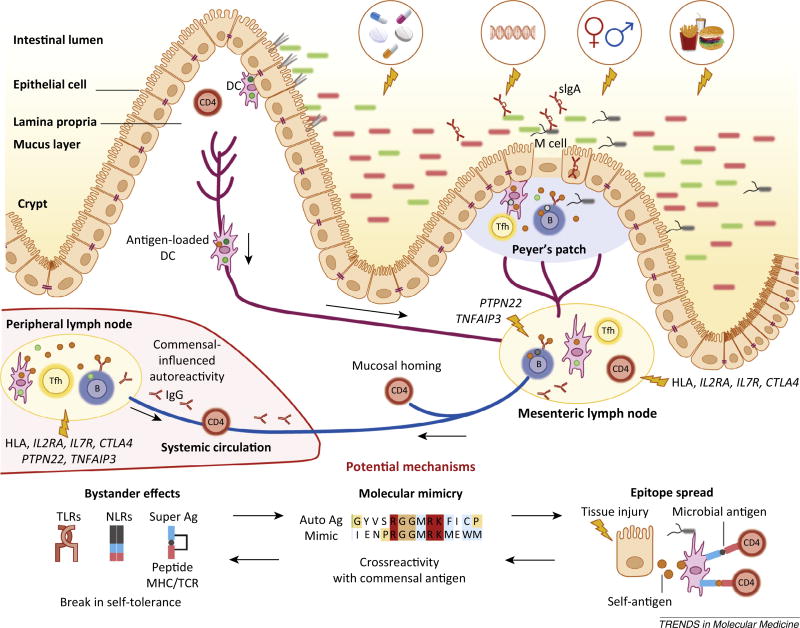
The microbiota is considered to be an important factor influencing the pathogenesis of autoimmunity at both barrier sites and internal organs. Impinging on innate and adaptive immunity, commensals exert protective or detrimental effects on various autoimmune animal models. Human microbiome studies of autoimmunity remain largely descriptive, but suggest a role for dysbiosis in autoimmune disease. Humanized gnotobiotic approaches have advanced our understanding of immune-commensal interactions, but little is known about the mechanisms in autoimmunity. We propose that, similarly to infectious agents, the microbiota mediates autoimmunity via bystander activation, epitope spread, and, particularly under homeostatic conditions, via crossreactivity. This review presents an overview of the current literature concluding with outstanding questions in this field.
Keywords: bystander activation; commensal; crossreactivity; epitope spread; microbiome; molecular mimicry.
Copyright © 2015 Elsevier Ltd. All rights reserved.
Figures
References
-
- Sommer F, Backhed F. The gut microbiota - masters of host development and physiology. Nat. Rev. Microbiol. 2013;11:227–238. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
