

Gene networks underlying convergent and pleiotropic phenotypes in a large and systematically-phenotyped cohort with heterogeneous developmental disorders
- PMID: 25781962
- PMCID: PMC4362763
- DOI: 10.1371/journal.pgen.1005012
Gene networks underlying convergent and pleiotropic phenotypes in a large and systematically-phenotyped cohort with heterogeneous developmental disorders
Abstract
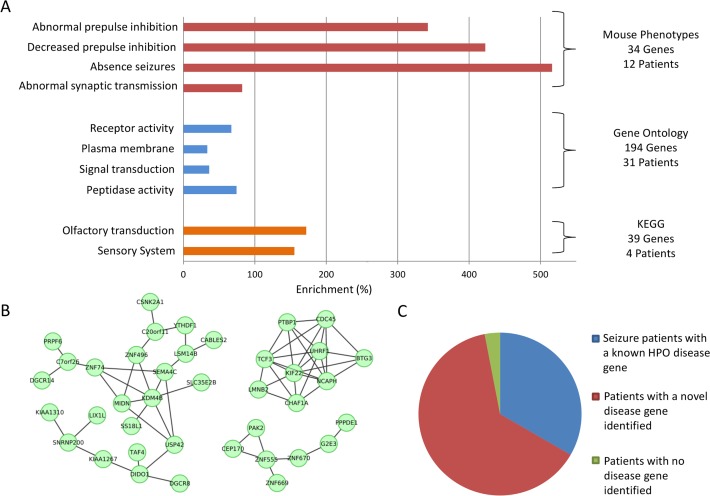
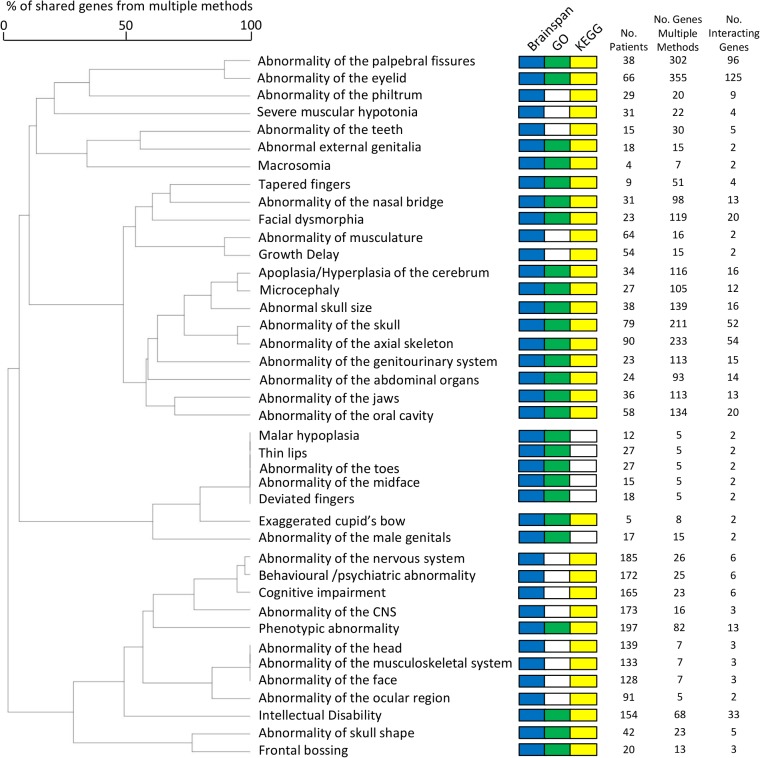
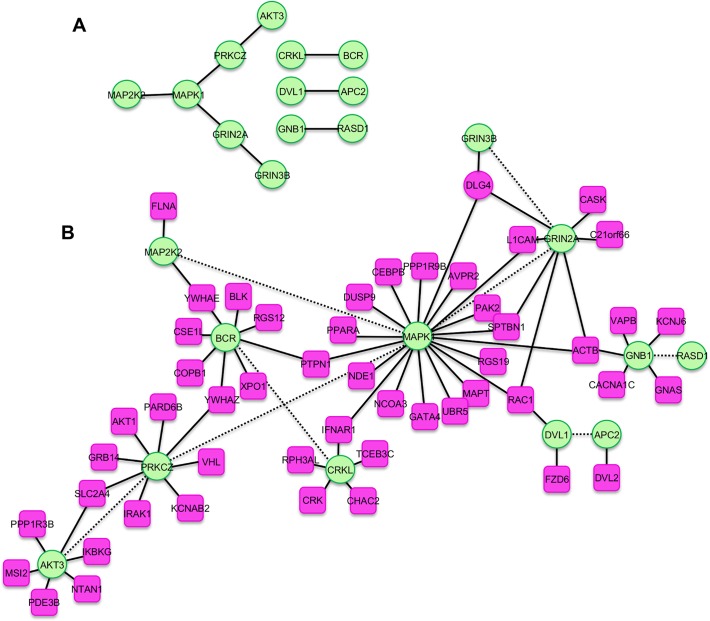
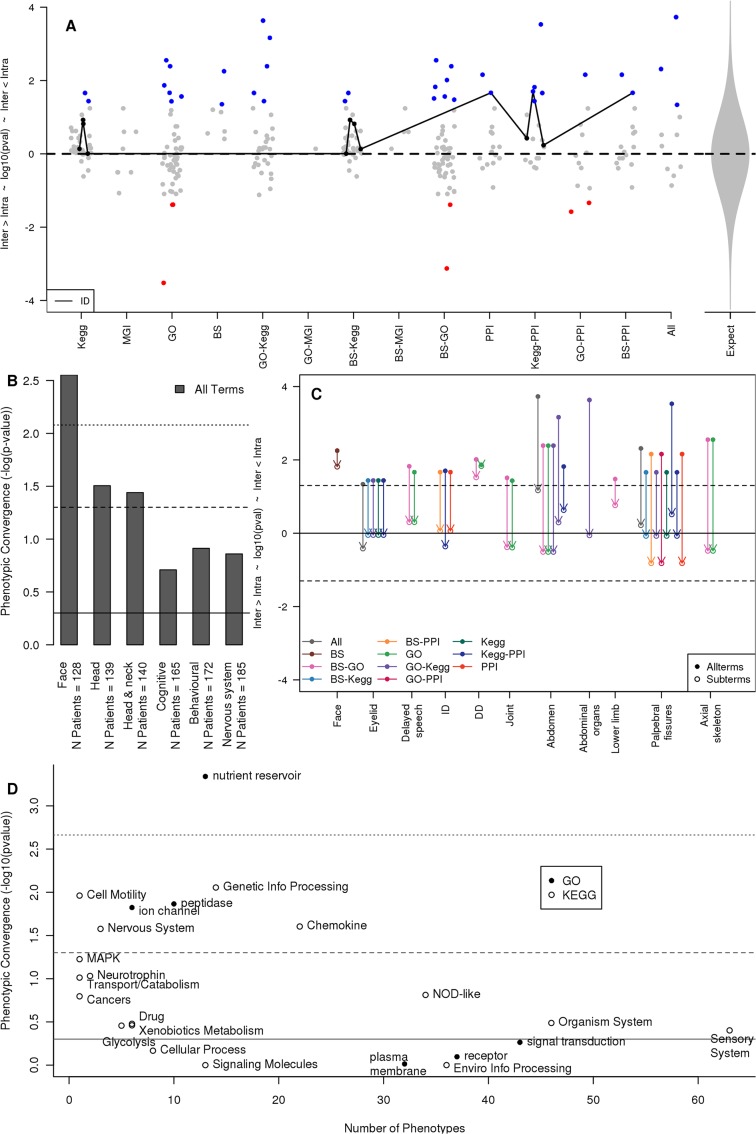
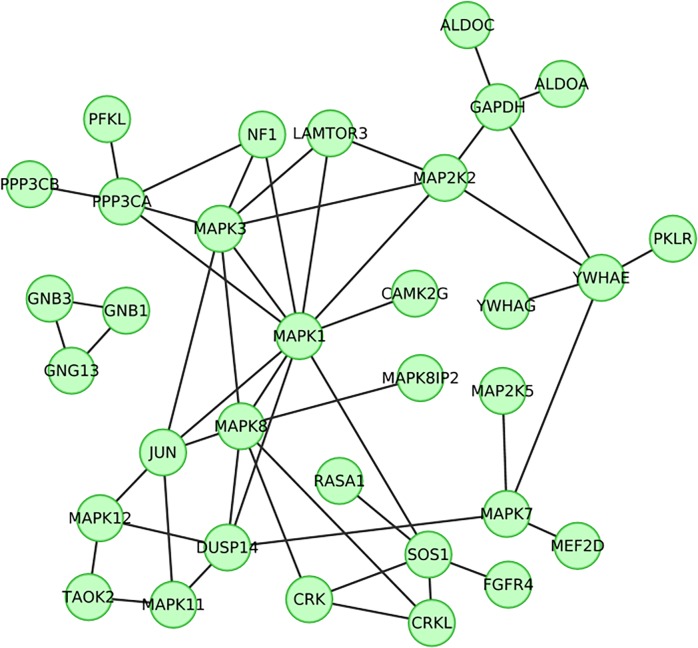
Readily-accessible and standardised capture of genotypic variation has revolutionised our understanding of the genetic contribution to disease. Unfortunately, the corresponding systematic capture of patient phenotypic variation needed to fully interpret the impact of genetic variation has lagged far behind. Exploiting deep and systematic phenotyping of a cohort of 197 patients presenting with heterogeneous developmental disorders and whose genomes harbour de novo CNVs, we systematically applied a range of commonly-used functional genomics approaches to identify the underlying molecular perturbations and their phenotypic impact. Grouping patients into 408 non-exclusive patient-phenotype groups, we identified a functional association amongst the genes disrupted in 209 (51%) groups. We find evidence for a significant number of molecular interactions amongst the association-contributing genes, including a single highly-interconnected network disrupted in 20% of patients with intellectual disability, and show using microcephaly how these molecular networks can be used as baits to identify additional members whose genes are variant in other patients with the same phenotype. Exploiting the systematic phenotyping of this cohort, we observe phenotypic concordance amongst patients whose variant genes contribute to the same functional association but note that (i) this relationship shows significant variation across the different approaches used to infer a commonly perturbed molecular pathway, and (ii) that the phenotypic similarities detected amongst patients who share the same inferred pathway perturbation result from these patients sharing many distinct phenotypes, rather than sharing a more specific phenotype, inferring that these pathways are best characterized by their pleiotropic effects.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
