

Genome diversity of Epstein-Barr virus from multiple tumor types and normal infection
- PMID: 25787276
- PMCID: PMC4442510
- DOI: 10.1128/JVI.03614-14
Genome diversity of Epstein-Barr virus from multiple tumor types and normal infection
Abstract
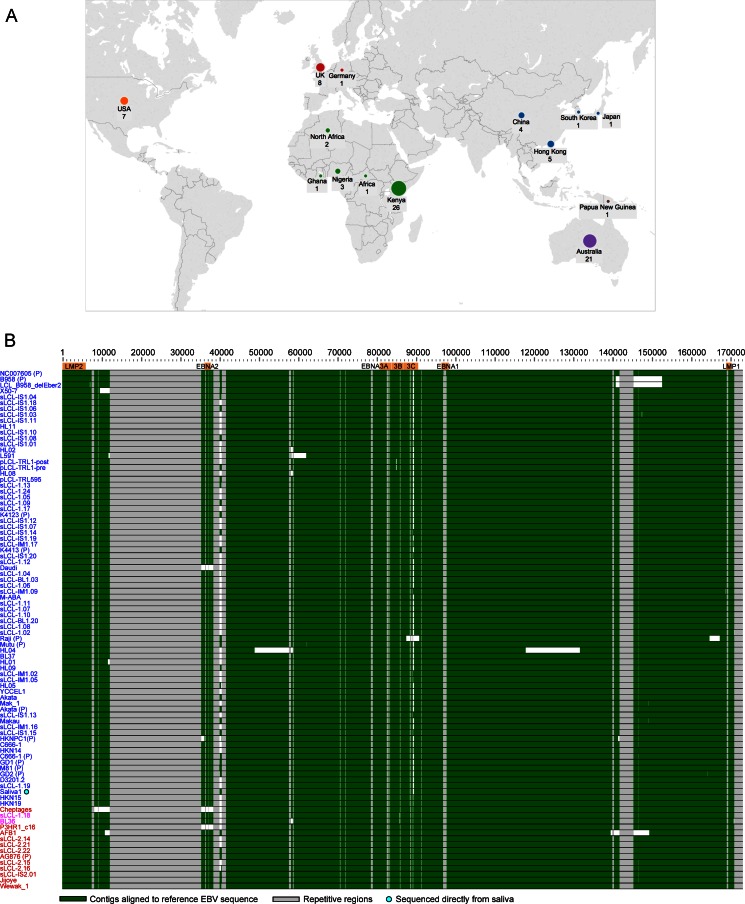
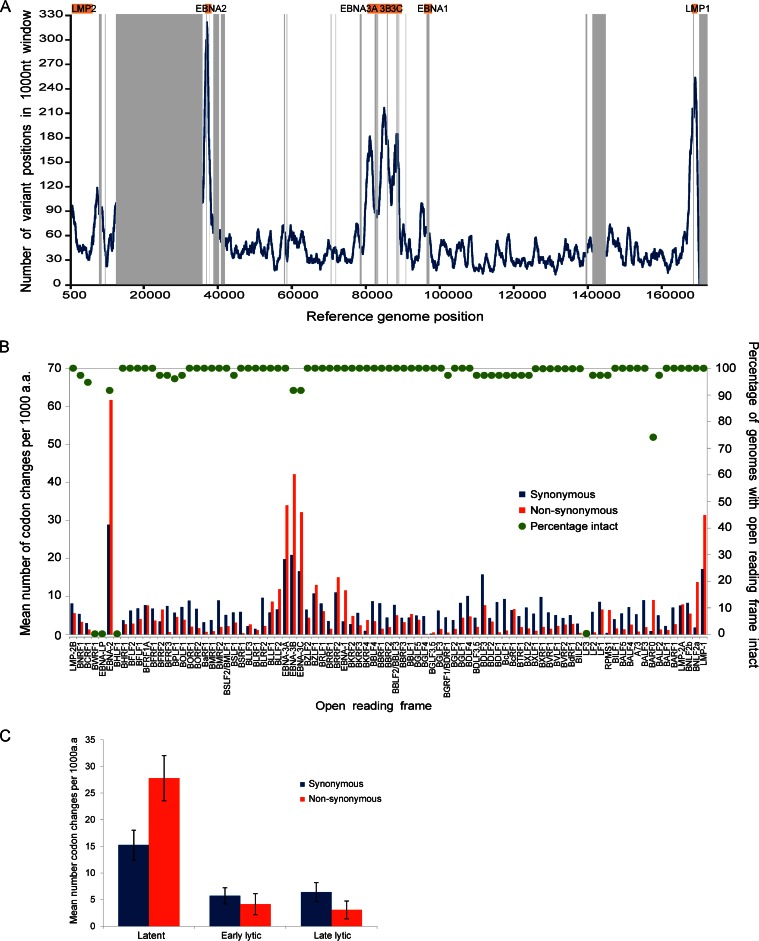
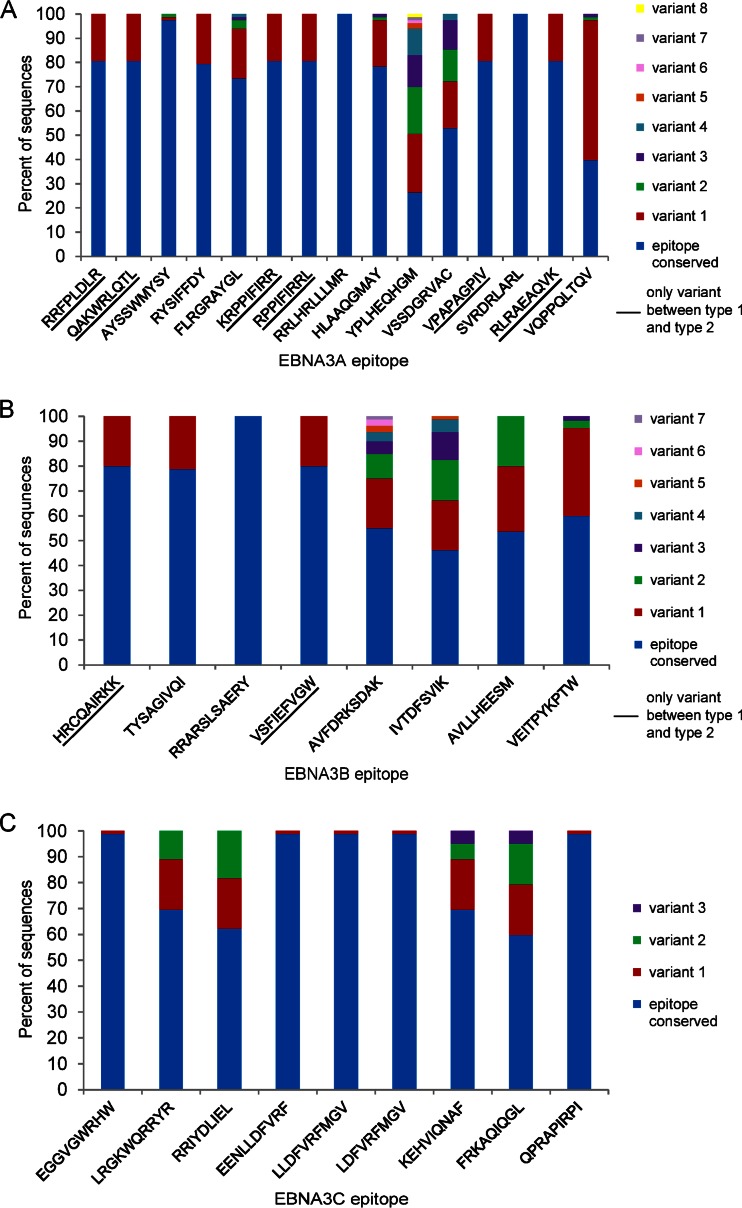
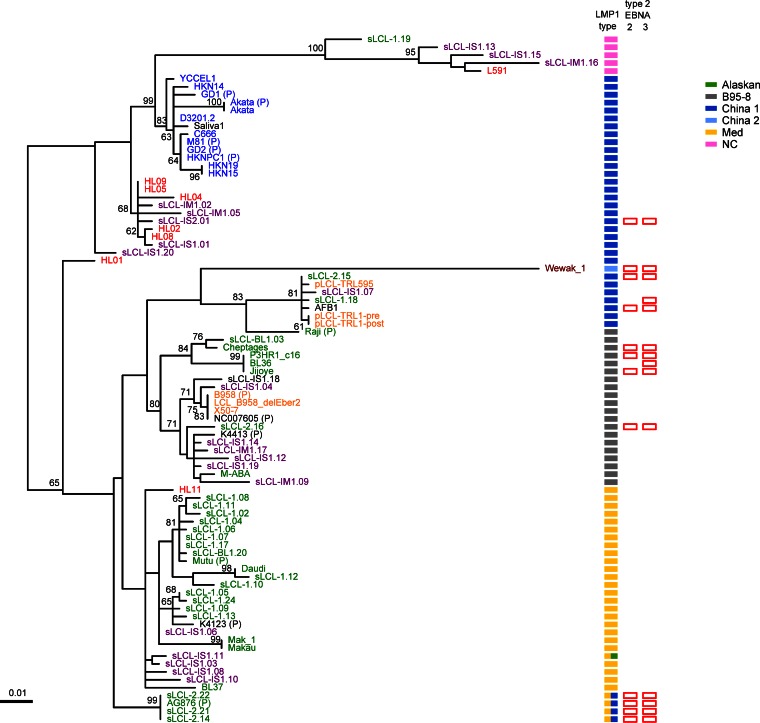
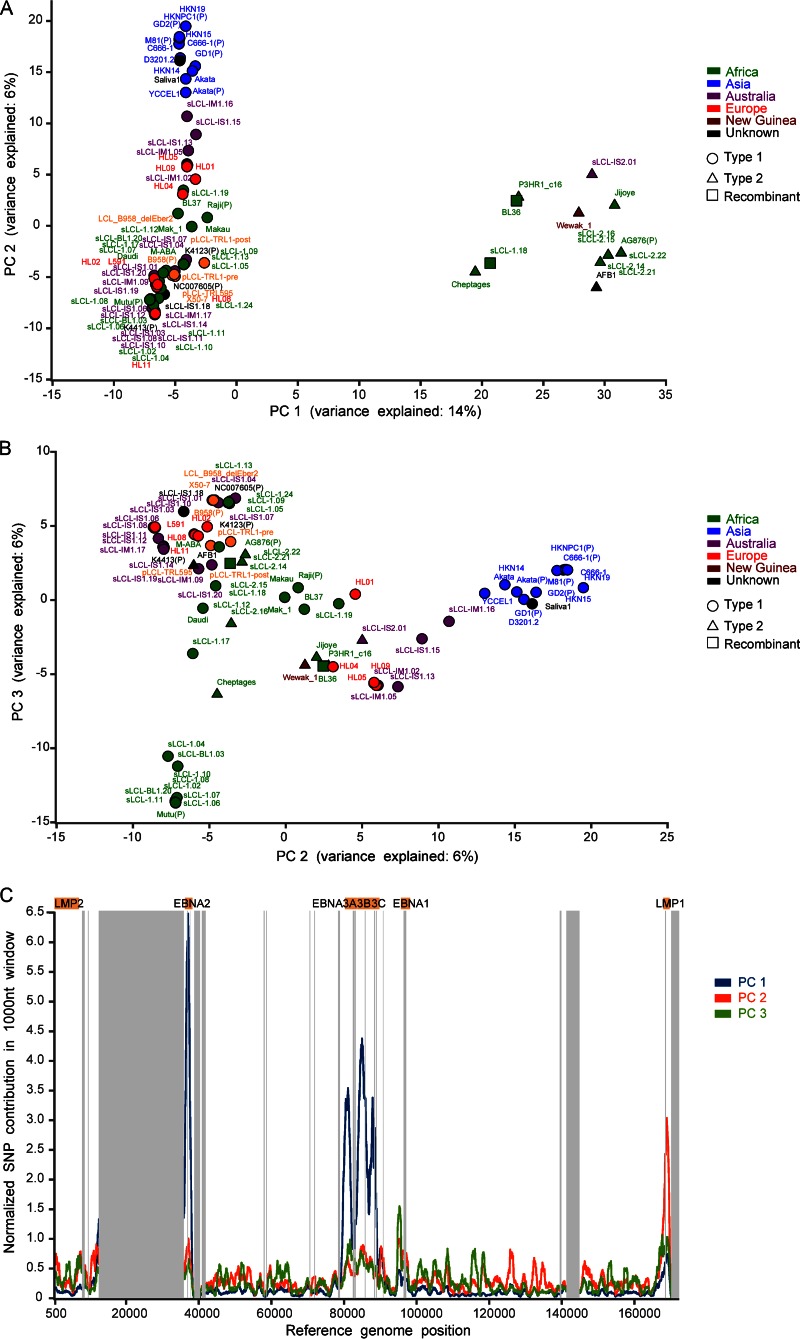
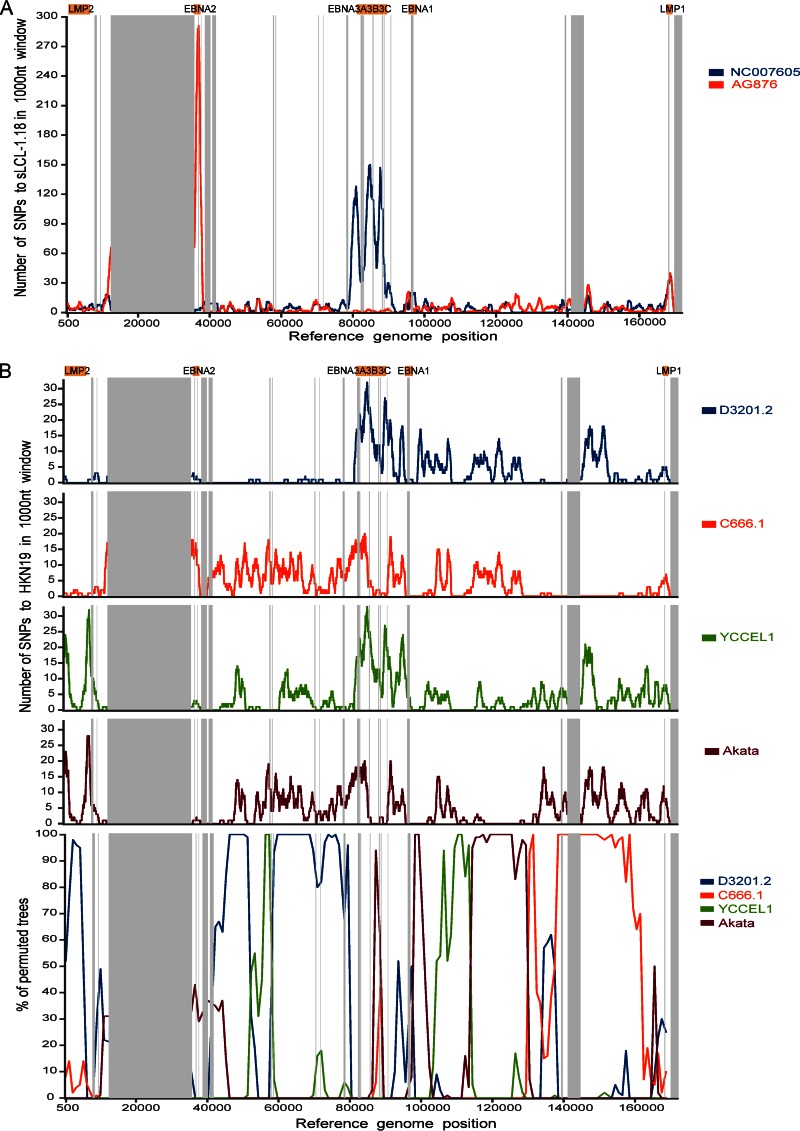
Epstein-Barr virus (EBV) infects most of the world's population and is causally associated with several human cancers, but little is known about how EBV genetic variation might influence infection or EBV-associated disease. There are currently no published wild-type EBV genome sequences from a healthy individual and very few genomes from EBV-associated diseases. We have sequenced 71 geographically distinct EBV strains from cell lines, multiple types of primary tumor, and blood samples and the first EBV genome from the saliva of a healthy carrier. We show that the established genome map of EBV accurately represents all strains sequenced, but novel deletions are present in a few isolates. We have increased the number of type 2 EBV genomes sequenced from one to 12 and establish that the type 1/type 2 classification is a major feature of EBV genome variation, defined almost exclusively by variation of EBNA2 and EBNA3 genes, but geographic variation is also present. Single nucleotide polymorphism (SNP) density varies substantially across all known open reading frames and is highest in latency-associated genes. Some T-cell epitope sequences in EBNA3 genes show extensive variation across strains, and we identify codons under positive selection, both important considerations for the development of vaccines and T-cell therapy. We also provide new evidence for recombination between strains, which provides a further mechanism for the generation of diversity. Our results provide the first global view of EBV sequence variation and demonstrate an effective method for sequencing large numbers of genomes to further understand the genetics of EBV infection.
Importance: Most people in the world are infected by Epstein-Barr virus (EBV), and it causes several human diseases, which occur at very different rates in different parts of the world and are linked to host immune system variation. Natural variation in EBV DNA sequence may be important for normal infection and for causing disease. Here we used rapid, cost-effective sequencing to determine 71 new EBV sequences from different sample types and locations worldwide. We showed geographic variation in EBV genomes and identified the most variable parts of the genome. We identified protein sequences that seem to have been selected by the host immune system and detected variability in known immune epitopes. This gives the first overview of EBV genome variation, important for designing vaccines and immune therapy for EBV, and provides techniques to investigate relationships between viral sequence variation and EBV-associated diseases.
Copyright © 2015, Palser et al.
Figures
References
-
- Kieff E, Rickinson A. 2007. Epstein-Barr virus and its replication, p 2603–2654. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA.
-
- White RE, Ramer PC, Naresh KN, Meixlsperger S, Pinaud L, Rooney C, Savoldo B, Coutinho R, Bodor C, Gribben J, Ibrahim HA, Bower M, Nourse JP, Gandhi MK, Middeldorp J, Cader FZ, Murray P, Munz C, Allday MJ. 2012. EBNA3B-deficient EBV promotes B cell lymphomagenesis in humanized mice and is found in human tumors. J Clin Invest 122:1487–1502. doi:10.1172/JCI58092. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
