

Mechanisms of neuroimmune gene induction in alcoholism
- PMID: 25787746
- PMCID: PMC4828484
- DOI: 10.1007/s00213-015-3906-1
Mechanisms of neuroimmune gene induction in alcoholism
Abstract
Rationale: Alcoholism is a primary, chronic relapsing disease of brain reward, motivation, memory, and related circuitry. It is characterized by an individual's continued drinking despite negative consequences related to alcohol use, which is exemplified by alcohol use leading to clinically significant impairment or distress. Chronic alcohol consumption increases the expression of innate immune signaling molecules (ISMs) in the brain that alter cognitive processes and promote alcohol drinking.
Objectives: Unraveling the mechanisms of alcohol-induced neuroimmune gene induction is complicated by positive loops of multiple cytokines and other signaling molecules that converge on nuclear factor kappa-light-chain-enhancer of activated B cells and activator protein-1 leading to induction of additional neuroimmune signaling molecules that amplify and expand the expression of ISMs.
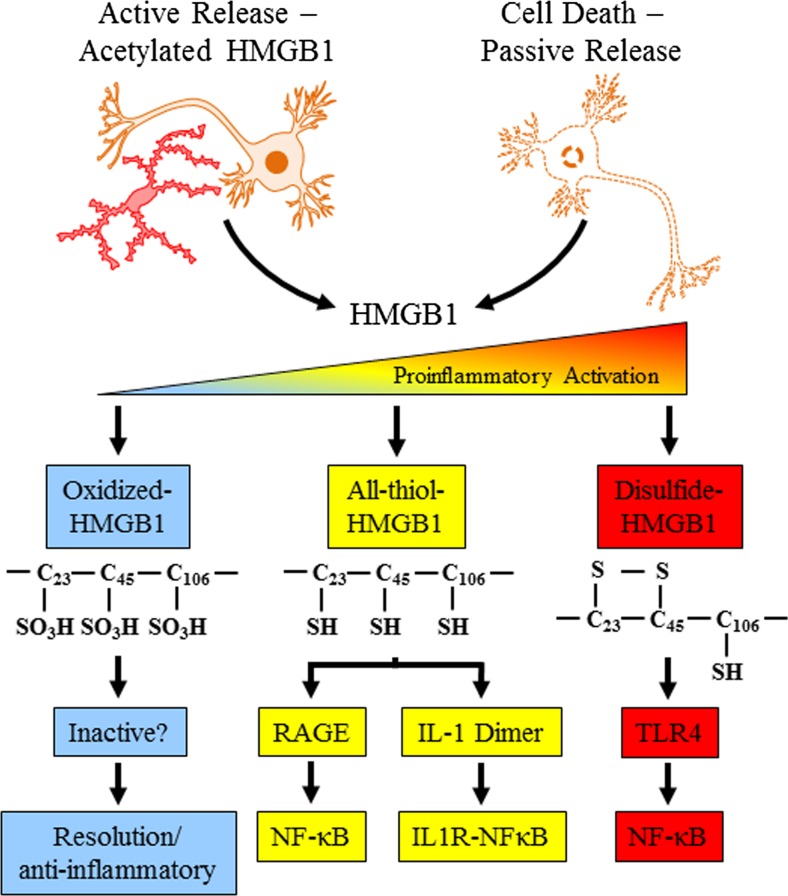
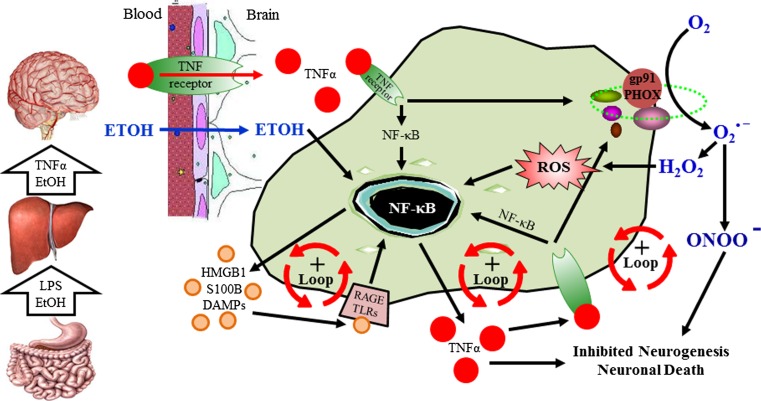
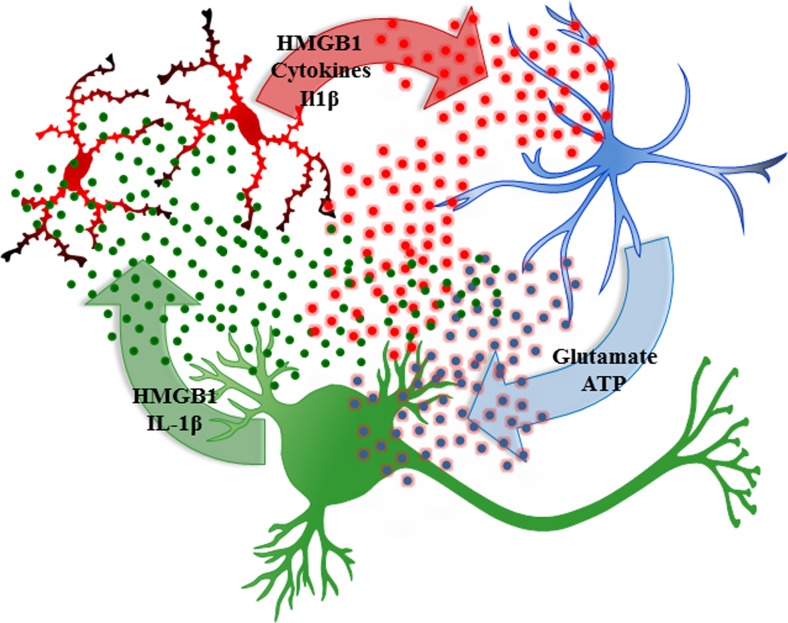
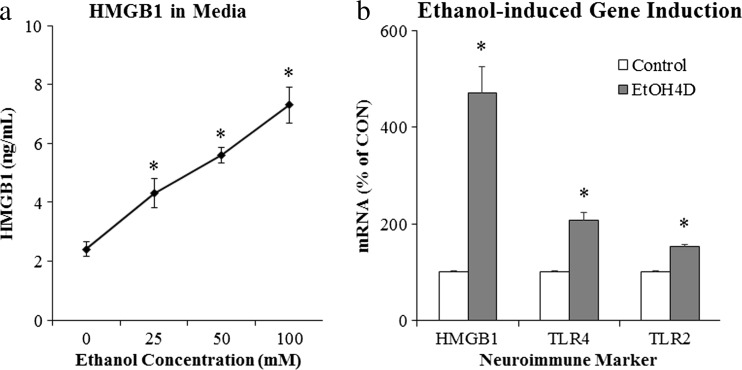
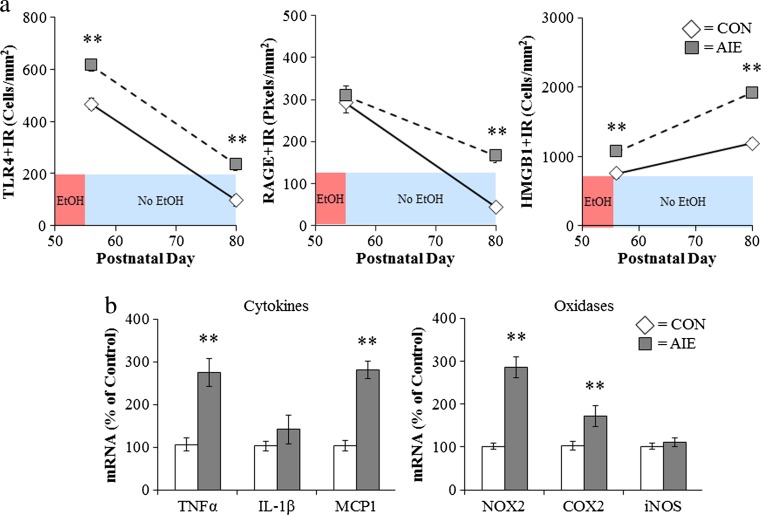
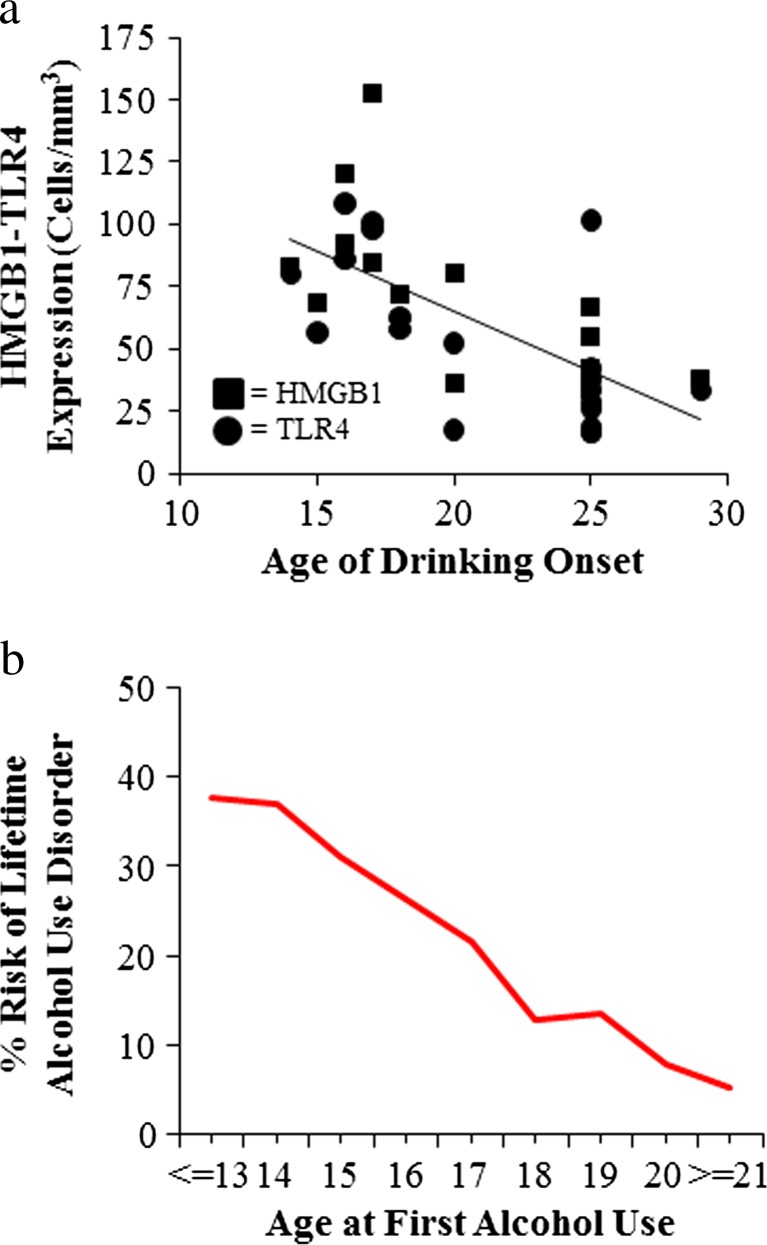
Results: Studies from our laboratory employing reverse transcription polymerase chain reaction (RT-PCR) to assess mRNA, immunohistochemistry and Western blot analysis to assess protein expression, and others suggest that ethanol increases brain neuroimmune gene and protein expression through two distinct mechanisms involving (1) systemic induction of innate immune molecules that are transported from blood to the brain and (2) the direct release of high-mobility group box 1 (HMGB1) from neurons in the brain. Released HMGB1 signals through multiple receptors, particularly Toll-like receptor (TLR) 4, that potentiate cytokine receptor responses leading to a hyperexcitable state that disrupts neuronal networks and increases excitotoxic neuronal death. Innate immune gene activation in brain is persistent, consistent with the chronic relapsing disease that is alcoholism. Expression of HMGB1, TLRs, and other ISMs is increased several-fold in the human orbital frontal cortex, and expression of these molecules is highly correlated with each other as well as lifetime alcohol consumption and age of drinking onset.
Conclusions: The persistent and cumulative nature of alcohol on HMGB1 and TLR gene induction support their involvement in alcohol-induced long-term changes in brain function and neurodegeneration.
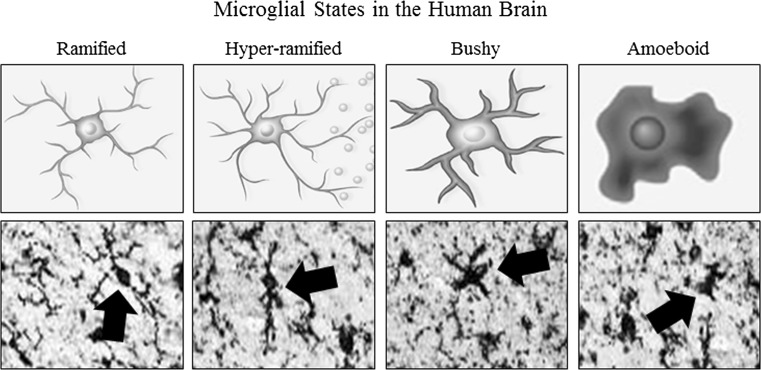
Keywords: Alcohol use disorder; Amphoterin; Cytokines; Ethanol; Frontal cortex; Gut permeability; HMGB1; Innate immune; Microglia; RAGE; TLR4.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- F32 AA021040/AA/NIAAA NIH HHS/United States
- AA007573/AA/NIAAA NIH HHS/United States
- P60 AA011605/AA/NIAAA NIH HHS/United States
- U24 AA020022/AA/NIAAA NIH HHS/United States
- L40 AA021590/AA/NIAAA NIH HHS/United States
- P50 AA011605/AA/NIAAA NIH HHS/United States
- U24 AA020024/AA/NIAAA NIH HHS/United States
- AA11605/AA/NIAAA NIH HHS/United States
- AA020022/AA/NIAAA NIH HHS/United States
- T32 AA007573/AA/NIAAA NIH HHS/United States
- AA019767/AA/NIAAA NIH HHS/United States
- AA020024/AA/NIAAA NIH HHS/United States
- U01 AA020023/AA/NIAAA NIH HHS/United States
- AA020023/AA/NIAAA NIH HHS/United States
- U54 AA019767/AA/NIAAA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
