

Dedicator of cytokinesis 2, a novel regulator for smooth muscle phenotypic modulation and vascular remodeling
- PMID: 25788409
- PMCID: PMC4426039
- DOI: 10.1161/CIRCRESAHA.116.305863
Dedicator of cytokinesis 2, a novel regulator for smooth muscle phenotypic modulation and vascular remodeling
Abstract
Rationale: Vascular smooth muscle cell (SMC) phenotypic modulation and vascular remodeling contribute to the development of several vascular disorders such as restenosis after angioplasty, transplant vasculopathy, and atherosclerosis. The mechanisms underlying these processes, however, remain largely unknown.
Objective: The objective of this study is to determine the role of dedicator of cytokinesis 2 (DOCK2) in SMC phenotypic modulation and vascular remodeling.
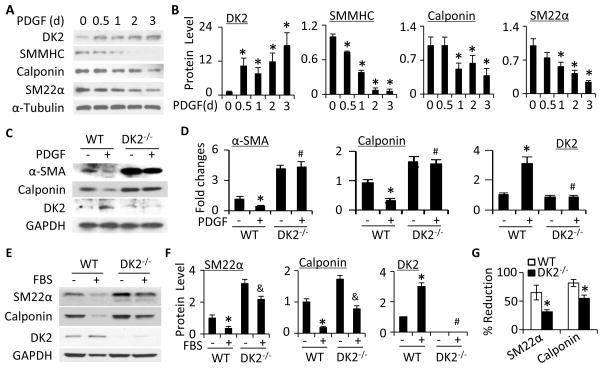
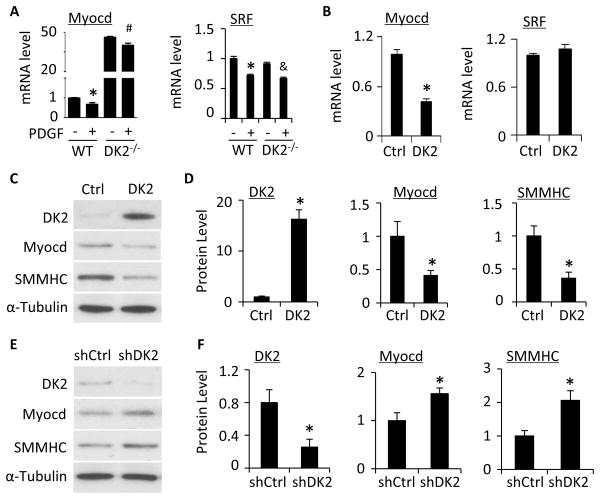
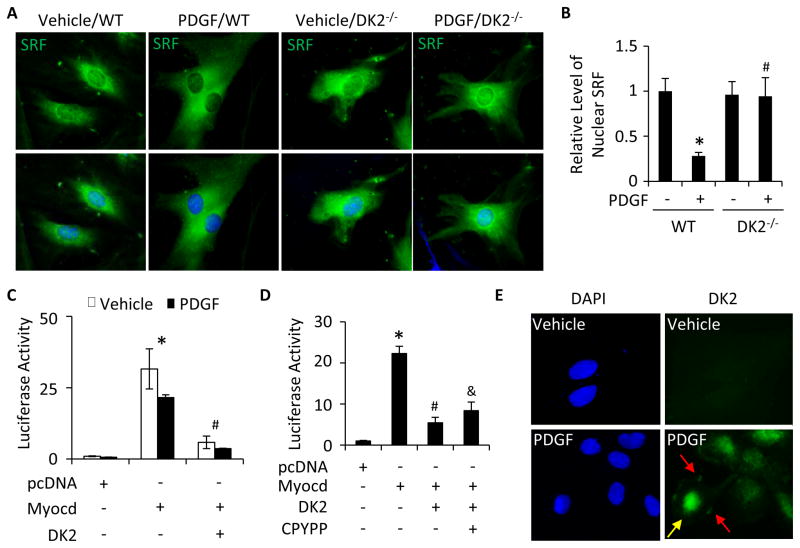
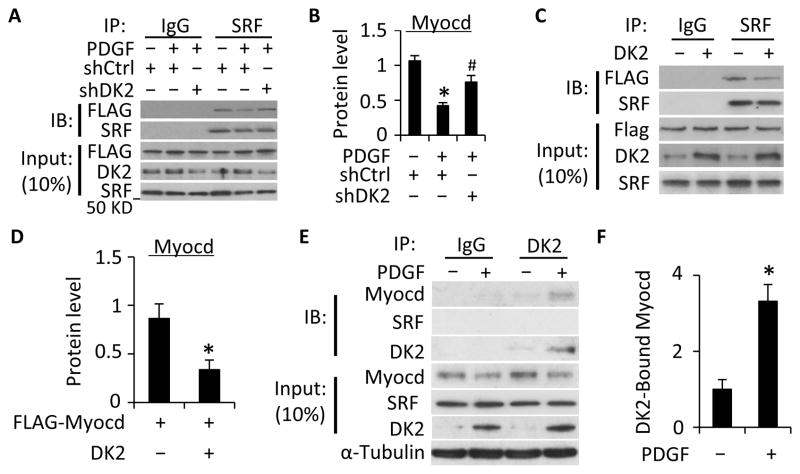
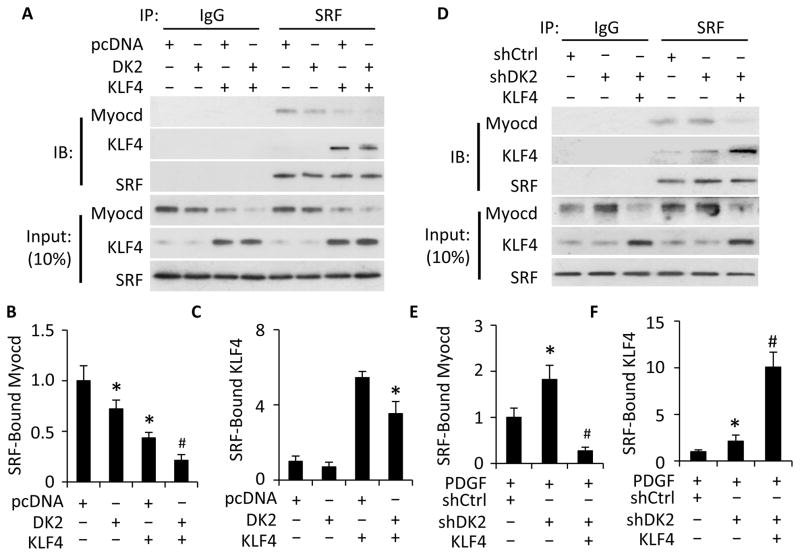
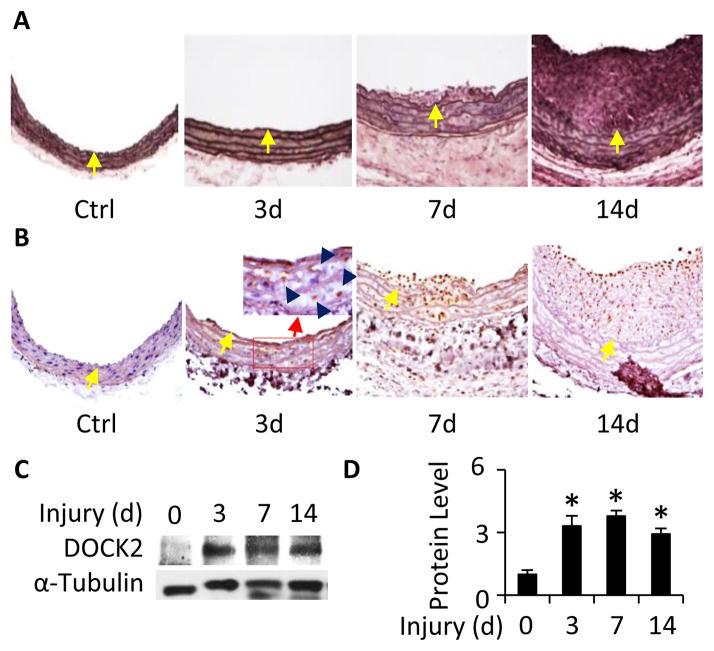
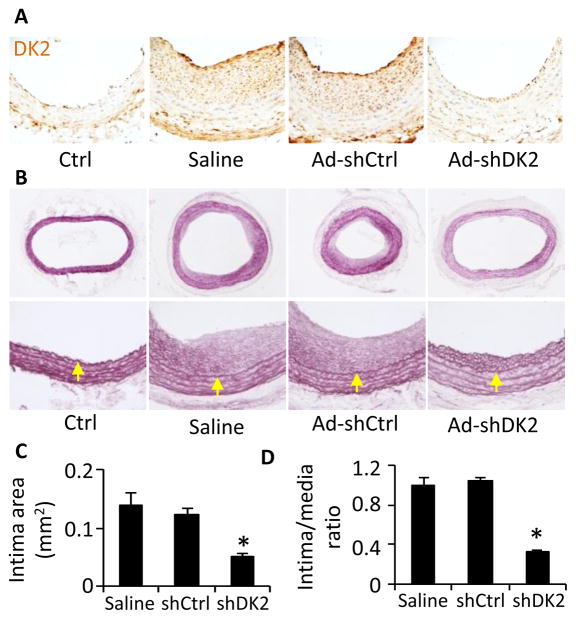
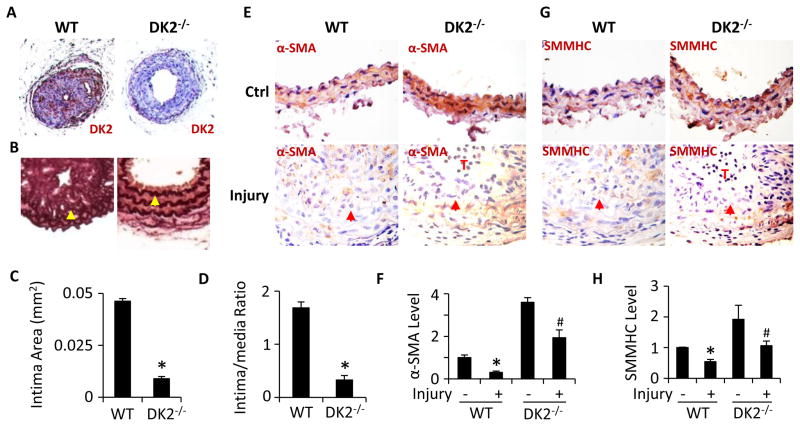
Methods and results: Platelet-derived growth factor-BB induced DOCK2 expression while modulating SMC phenotype. DOCK2 deficiency diminishes platelet-derived growth factor-BB or serum-induced downregulation of SMC markers. Conversely, DOCK2 overexpression inhibits SMC marker expression in primary cultured SMC. Mechanistically, DOCK2 inhibits myocardin expression, blocks serum response factor nuclear location, attenuates myocardin binding to serum response factor, and thus attenuates myocardin-induced smooth muscle marker promoter activity. Moreover, DOCK2 and Kruppel-like factor 4 cooperatively inhibit myocardin-serum response factor interaction. In a rat carotid artery balloon-injury model, DOCK2 is induced in media layer SMC initially and neointima SMC subsequently after vascular injury. Knockdown of DOCK2 dramatically inhibits the neointima formation by 60%. Most importantly, knockout of DOCK2 in mice markedly blocks ligation-induced intimal hyperplasia while restoring SMC contractile protein expression.
Conclusions: Our studies identified DOCK2 as a novel regulator for SMC phenotypic modulation and vascular lesion formation after vascular injury. Therefore, targeting DOCK2 may be a potential therapeutic strategy for the prevention of vascular remodeling in proliferative vascular diseases.
Keywords: cell proliferation; dedicator of cytokinesis 2; vascular remodeling.
© 2015 American Heart Association, Inc.
Figures
References
-
- Schwartz SM, deBlois D, O’Brien ER. The intima. Soil for atherosclerosis and restenosis. Circulation research. 1995;77:445–465. - PubMed
-
- Neitzel GF, Barboriak JJ, Pintar K, Qureshi I. Atherosclerosis in aortocoronary bypass grafts. Morphologic study and risk factor analysis 6 to 12 years after surgery. Arteriosclerosis. 1986;6:594–600. - PubMed
-
- Schwartz SM. Smooth muscle migration in atherosclerosis and restenosis. The Journal of clinical investigation. 1997;100:S87–89. - PubMed
-
- Dzau VJ, Braun-Dullaeus RC, Sedding DG. Vascular proliferation and atherosclerosis: New perspectives and therapeutic strategies. Nature medicine. 2002;8:1249–1256. - PubMed
-
- Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiological reviews. 2004;84:767–801. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
