

Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia
- PMID: 25790293
- PMCID: PMC4377644
- DOI: 10.1038/ncomms7604
Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia
Abstract
There is incomplete understanding of genetic heterogeneity and clonal evolution during cancer progression. Here we use deep whole-exome sequencing to describe the clonal architecture and evolution of 20 pediatric B-acute lymphoblastic leukaemias from diagnosis to relapse. We show that clonal diversity is comparable at diagnosis and relapse and clonal survival from diagnosis to relapse is not associated with mutation burden. Six pathways were frequently mutated, with NT5C2, CREBBP, WHSC1, TP53, USH2A, NRAS and IKZF1 mutations enriched at relapse. Half of the leukaemias had multiple subclonal mutations in a pathway or gene at diagnosis, but mostly with only one, usually minor clone, surviving therapy to acquire additional mutations and become the relapse founder clone. Relapse-specific mutations in NT5C2 were found in nine cases, with mutations in four cases being in descendants of the relapse founder clone. These results provide important insights into the genetic basis of treatment failure in ALL and have implications for the early detection of mutations driving relapse.
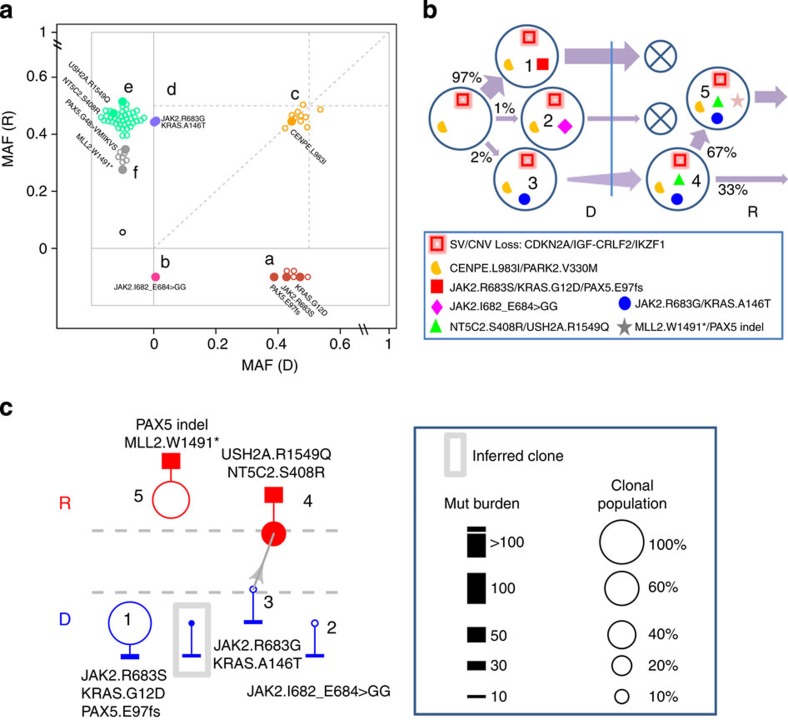
Figures





References
-
- Place A. E., Frederick N. N. & Sallan S. E. Therapeutic approaches to haematological malignancies in adolescents and young adults. Br. J. Haematol. 164, 3–14 (2014) . - PubMed
-
- Schrappe M. et al. Key treatment questions in childhood acute lymphoblastic leukemia: results in 5 consecutive trials performed by the ALL-BFM study group from 1981 to 2000. Klin. Padiatr. 225, (Suppl 1): S62–S72 (2013) . - PubMed
-
- Vora A. et al. Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): a randomised controlled trial. Lancet Oncol. 14, 199–209 (2013) . - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
- GM92666/GM/NIGMS NIH HHS/United States
- U10 CA98413/CA/NCI NIH HHS/United States
- P30 CA021765/CA/NCI NIH HHS/United States
- U01 CA157937/CA/NCI NIH HHS/United States
- U10 CA098543/CA/NCI NIH HHS/United States
- HHSN261200800001C/RC/CCR NIH HHS/United States
- U10 CA180886/CA/NCI NIH HHS/United States
- U01 GM092666/GM/NIGMS NIH HHS/United States
- U10 CA098413/CA/NCI NIH HHS/United States
- U24 CA114766/CA/NCI NIH HHS/United States
- CA21765/CA/NCI NIH HHS/United States
- R01 CA161202/CA/NCI NIH HHS/United States
- HHSN261200800001E/CA/NCI NIH HHS/United States
- U10 CA98543/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
