

Sequencing and analysis of globally obtained human respiratory syncytial virus A and B genomes
- PMID: 25793751
- PMCID: PMC4368745
- DOI: 10.1371/journal.pone.0120098
Sequencing and analysis of globally obtained human respiratory syncytial virus A and B genomes
Abstract
Background: Human respiratory syncytial virus (RSV) is the leading cause of respiratory tract infections in children globally, with nearly all children experiencing at least one infection by the age of two. Partial sequencing of the attachment glycoprotein gene is conducted routinely for genotyping, but relatively few whole genome sequences are available for RSV. The goal of our study was to sequence the genomes of RSV strains collected from multiple countries to further understand the global diversity of RSV at a whole-genome level.
Methods: We collected RSV samples and isolates from Mexico, Argentina, Belgium, Italy, Germany, Australia, South Africa, and the USA from the years 1998-2010. Both Sanger and next-generation sequencing with the Illumina and 454 platforms were used to sequence the whole genomes of RSV A and B. Phylogenetic analyses were performed using the Bayesian and maximum likelihood methods of phylogenetic inference.
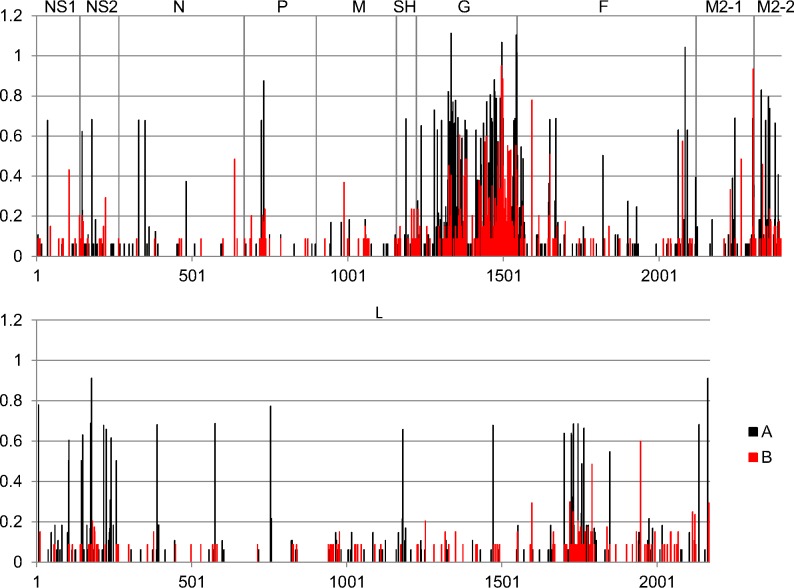
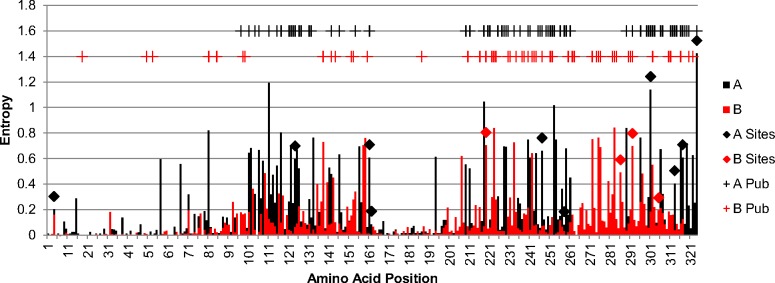
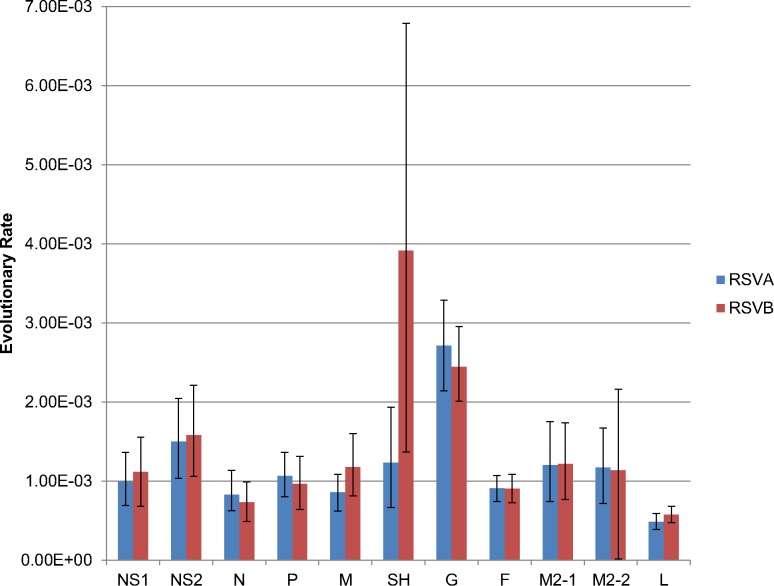
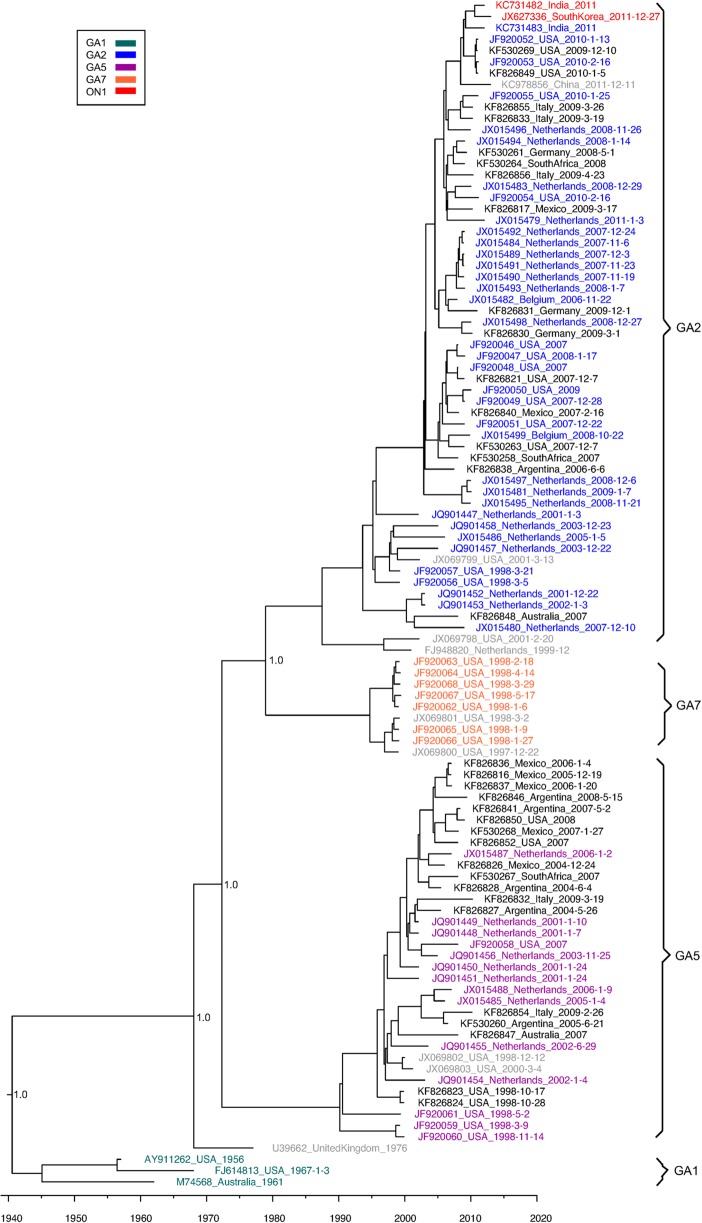
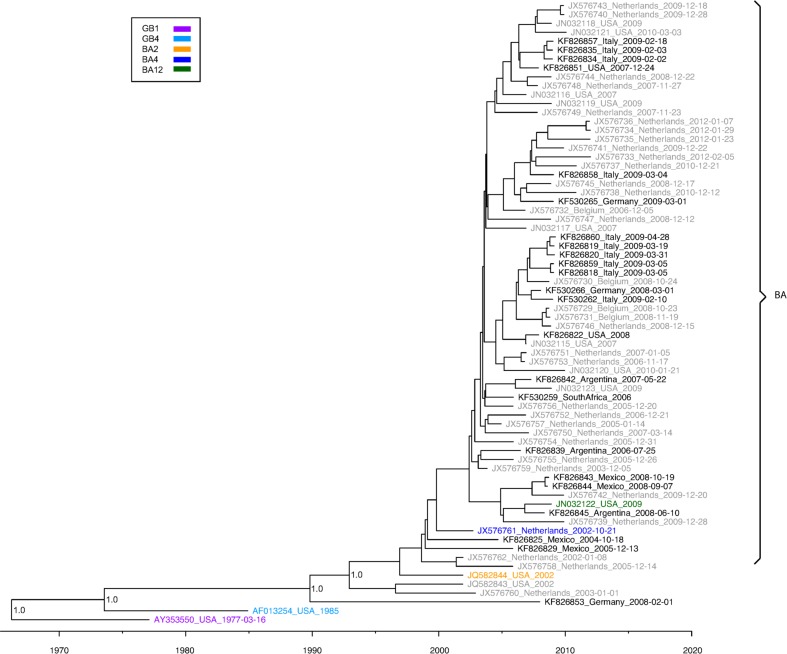
Results: We sequenced the genomes of 34 RSVA and 23 RSVB viruses. Phylogenetic analysis showed that the RSVA genome evolves at an estimated rate of 6.72 × 10(-4) substitutions/site/year (95% HPD 5.61 × 10(-4) to 7.6 × 10(-4)) and for RSVB the evolutionary rate was 7.69 × 10(-4) substitutions/site/year (95% HPD 6.81 × 10(-4) to 8.62 × 10(-4)). We found multiple clades co-circulating globally for both RSV A and B. The predominant clades were GA2 and GA5 for RSVA and BA for RSVB.
Conclusions: Our analyses showed that RSV circulates on a global scale with the same predominant clades of viruses being found in countries around the world. However, the distribution of clades can change rapidly as new strains emerge. We did not observe a strong spatial structure in our trees, with the same three main clades of RSV co-circulating globally, suggesting that the evolution of RSV is not strongly regionalized.
Conflict of interest statement
Figures
References
-
- Glezen WP, Taber LH, Frank AL, Kasel JA. Risk of primary infection and reinfection with respiratory syncytial virus. Am J Dis Child. 1986;140(6):543–6. - PubMed
-
- Falsey AR. Respiratory syncytial virus infection in elderly and high-risk adults. Exp Lung Res. 2005;31 Suppl 1:77 - PubMed
-
- Sorvillo FJ, Huie SF, Strassburg MA, Butsumyo A, Shandera WX, Fannin SL. An outbreak of respiratory syncytial virus pneumonia in a nursing home for the elderly. J Infect. 1984;9(3):252–6. - PubMed
-
- Mufson MA, Orvell C, Rafnar B, Norrby E. Two distinct subtypes of human respiratory syncytial virus. J Gen Virol. 1985;66 (Pt 10):2111–24. - PubMed
-
- Cane PA, Matthews DA, Pringle CR. Identification of variable domains of the attachment (G) protein of subgroup A respiratory syncytial viruses. J Gen Virol. 1991;72 (Pt 9):2091–6. - PubMed
Publication types
MeSH terms
Associated data
- BioProject/PRJNA73049
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
