

Glutamate and GABA imbalance following traumatic brain injury
- PMID: 25796572
- PMCID: PMC4640931
- DOI: 10.1007/s11910-015-0545-1
Glutamate and GABA imbalance following traumatic brain injury
Abstract
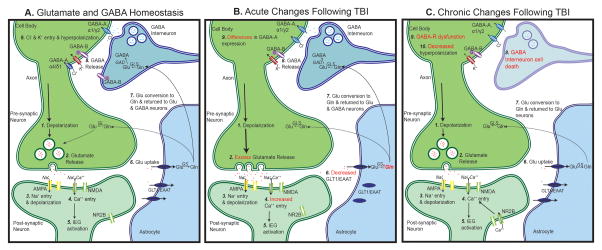
Traumatic brain injury (TBI) leads to multiple short- and long-term changes in neuronal circuits that ultimately conclude with an imbalance of cortical excitation and inhibition. Changes in neurotransmitter concentrations, receptor populations, and specific cell survival are important contributing factors. Many of these changes occur gradually, which may explain the vulnerability of the brain to multiple mild impacts, alterations in neuroplasticity, and delays in the presentation of posttraumatic epilepsy. In this review, we provide an overview of normal glutamate and GABA homeostasis and describe acute, subacute, and chronic changes that follow injury. We conclude by highlighting opportunities for therapeutic interventions in this paradigm.
Figures
References
-
- Spruston N. Pyramidal neurons: dendritic structure and synaptic integration. Nature Reviews Neuroscience. 2008 Mar;9(3):206–21. - PubMed
-
- Kandel E. In: Principles of Neural Science. 5. Kandel ER, Schwartz JH, Jessell TM, Siegelbaum SA, Hudspeth AJ, editors. McGraw Hill Professional; 2013. pp. 210–306.
-
- Castro-Alamancos MA, Connors BW. Thalamocortical synapses. Progress in Neurobiology. 1997 Apr;51(6):581–606. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
