

One cannot rule them all: Are bacterial toxins-antitoxins druggable?
- PMID: 25796610
- PMCID: PMC4487406
- DOI: 10.1093/femsre/fuv002
One cannot rule them all: Are bacterial toxins-antitoxins druggable?
Abstract
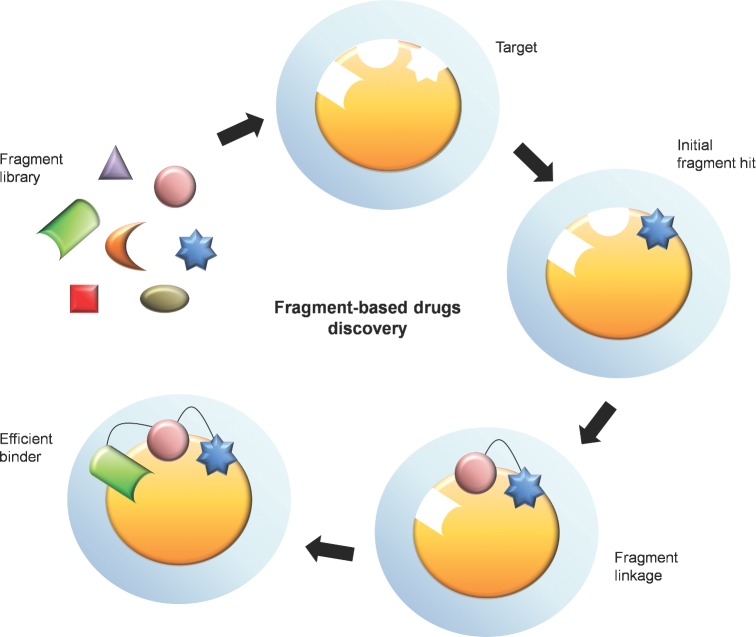
Type II (proteic) toxin-antitoxin (TA) operons are widely spread in bacteria and archaea. They are organized as operons in which, usually, the antitoxin gene precedes the cognate toxin gene. The antitoxin generally acts as a transcriptional self-repressor, whereas the toxin acts as a co-repressor, both proteins constituting a harmless complex. When bacteria encounter a stressful environment, TAs are triggered. The antitoxin protein is unstable and will be degraded by host proteases, releasing the free toxin to halt essential processes. The result is a cessation of cell growth or even death. Because of their ubiquity and the essential processes targeted, TAs have been proposed as good candidates for development of novel antimicrobials. We discuss here the possible druggability of TAs as antivirals and antibacterials, with focus on the potentials and the challenges that their use may find in the 'real' world. We present strategies to develop TAs as antibacterials in view of novel technologies, such as the use of very small molecules (fragments) as inhibitors of protein-protein interactions. Appropriate fragments could disrupt the T:A interfaces leading to the release of the targeted TA pair. Possible ways of delivery and formulation of Tas are also discussed.
Keywords: antibacterials; antivirals; drug delivery; drug discovery; inhibitors of protein–protein interactions; persistence; toxin–antitoxin operons.
© FEMS 2015.
Figures






References
-
- Alonso JC, Balsa D, Cherny I, et al. Bacterial toxin-antitoxin systems as targets for the development of novel antibiotics. In: Bonomo RA, Tolmasky ME, et al., editors. Enzyme-Mediated Resistance to Antibiotics: Mechanisms, Dissemination, and Prospects for Inhibition. Washington, DC: ASM Press; 2007. pp. 313–29.
-
- Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2012;303:1818–21. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
