

Emerging technologies for studying DNA methylation for the molecular diagnosis of cancer
- PMID: 25797072
- PMCID: PMC4422757
- DOI: 10.1586/14737159.2015.1027194
Emerging technologies for studying DNA methylation for the molecular diagnosis of cancer
Abstract
DNA methylation is an epigenetic mechanism that plays a key role in regulating gene expression and other functions. Although this modification is seen in different sequence contexts, the most frequently detected DNA methylation in mammals involves cytosine-guanine dinucleotides. Pathological alterations in DNA methylation patterns are described in a variety of human diseases, including cancer. Unlike genetic changes, DNA methylation is heavily influenced by subtle modifications in the cellular microenvironment. In all cancers, aberrant DNA methylation is involved in the alteration of a large number of oncological pathways with relevant theranostic utility. Several technologies for DNA methylation mapping have been developed recently and successfully applied in cancer studies. The scope of these technologies varies from assessing a single cytosine-guanine locus to genome-wide distribution of DNA methylation. Here, we review the strengths and weaknesses of these approaches in the context of clinical utility for the molecular diagnosis of human cancers.
Keywords: DNA methylation; cancer diagnosis; epigenomics; microarray; sequencing.
Conflict of interest statement
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Figures
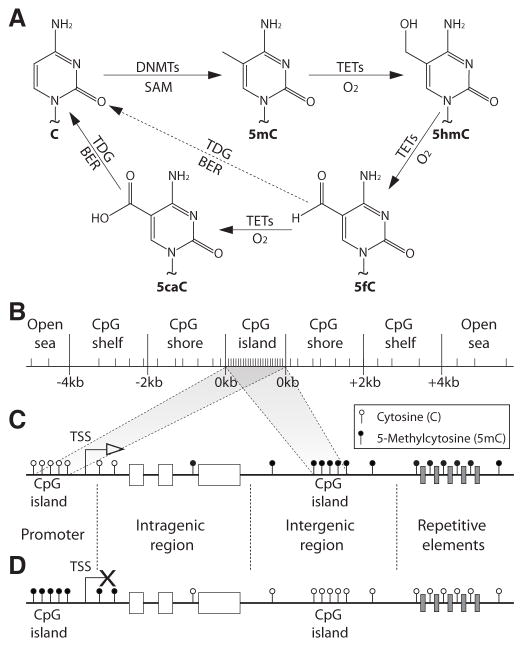
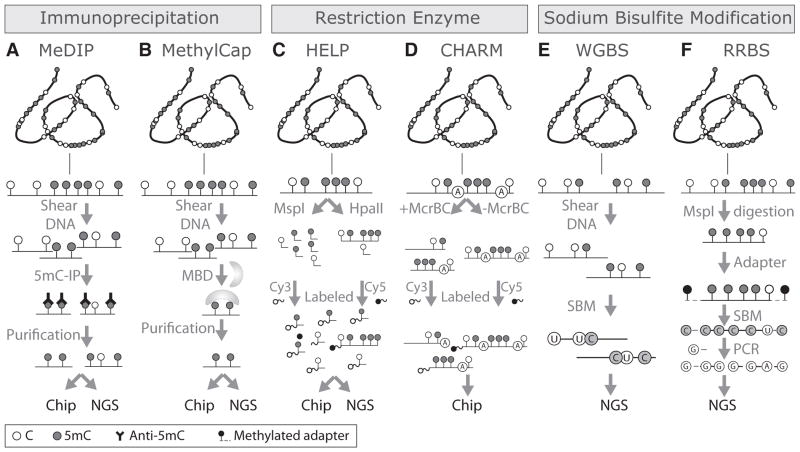
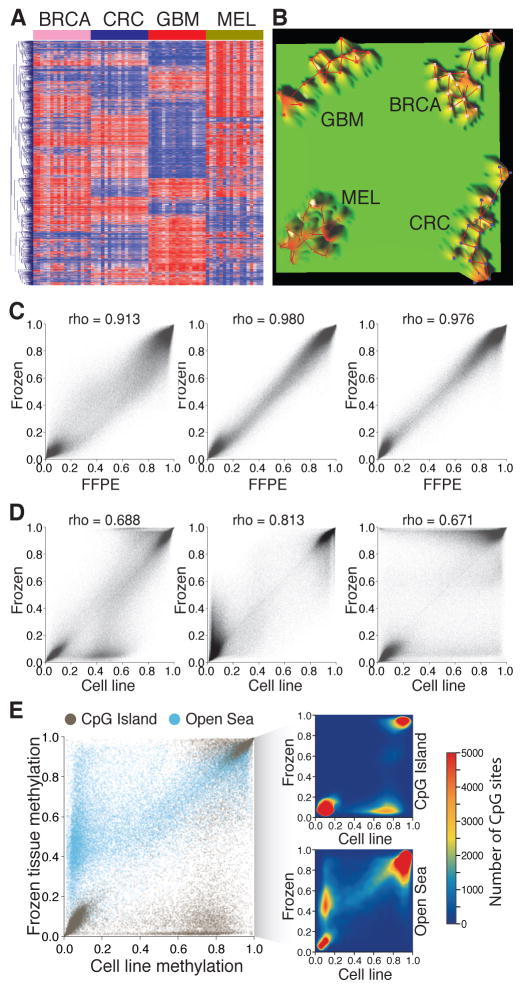
References
-
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature genetics. 2003;33 (Suppl):245–254. - PubMed
-
- Peters J. The role of genomic imprinting in biology and disease: an expanding view. Nat Rev Genet. 2014;15(8):517–530. - PubMed
-
- Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14(3):204–220. - PubMed
-
- Lister R, Pelizzola M, Dowen RH, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462(7271):315–322. This study was the first whole-genome DNA methylation mapping of a human genome and describes the presence of non-CpG dinucleotide methylation. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources