

Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment
- PMID: 25798620
- PMCID: PMC4396477
- DOI: 10.1172/JCI78239
Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment
Abstract
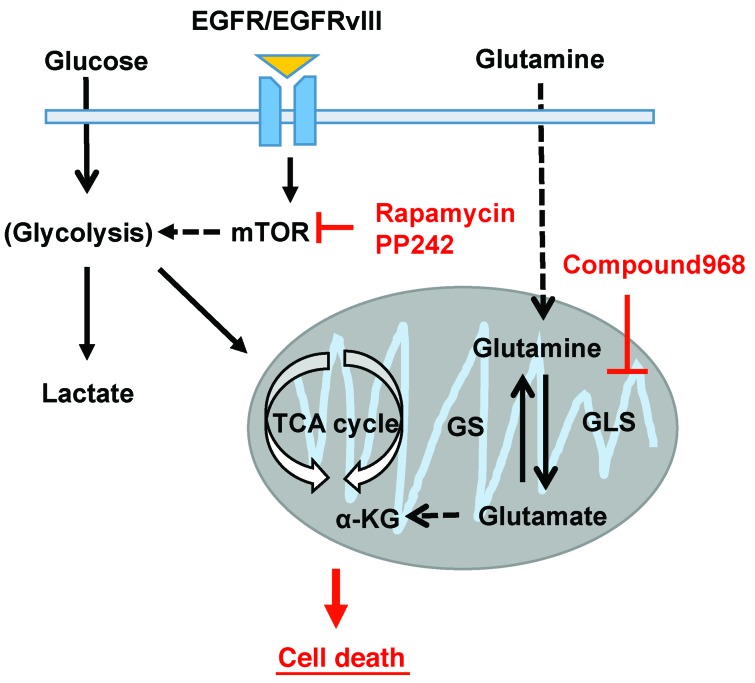
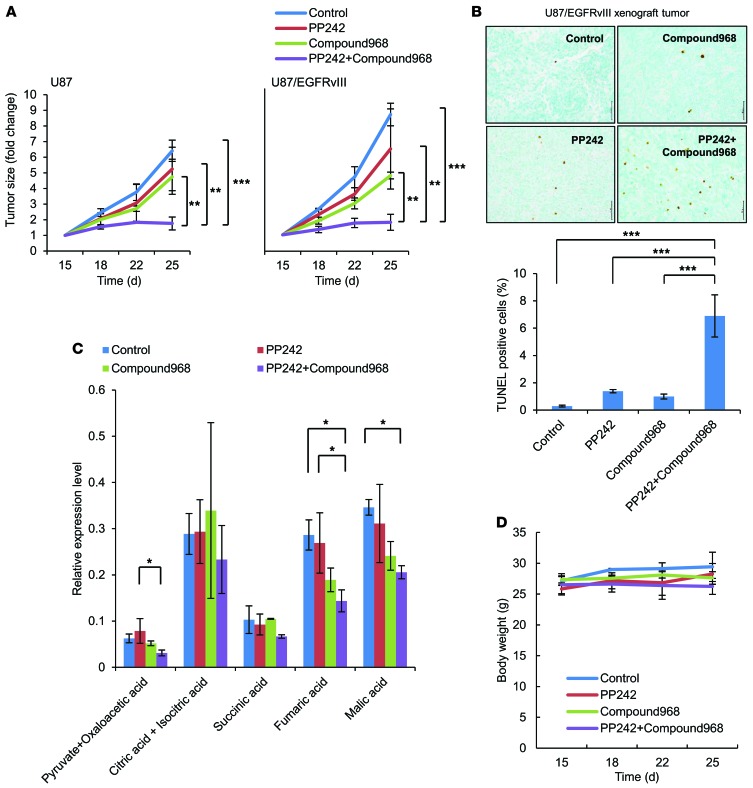
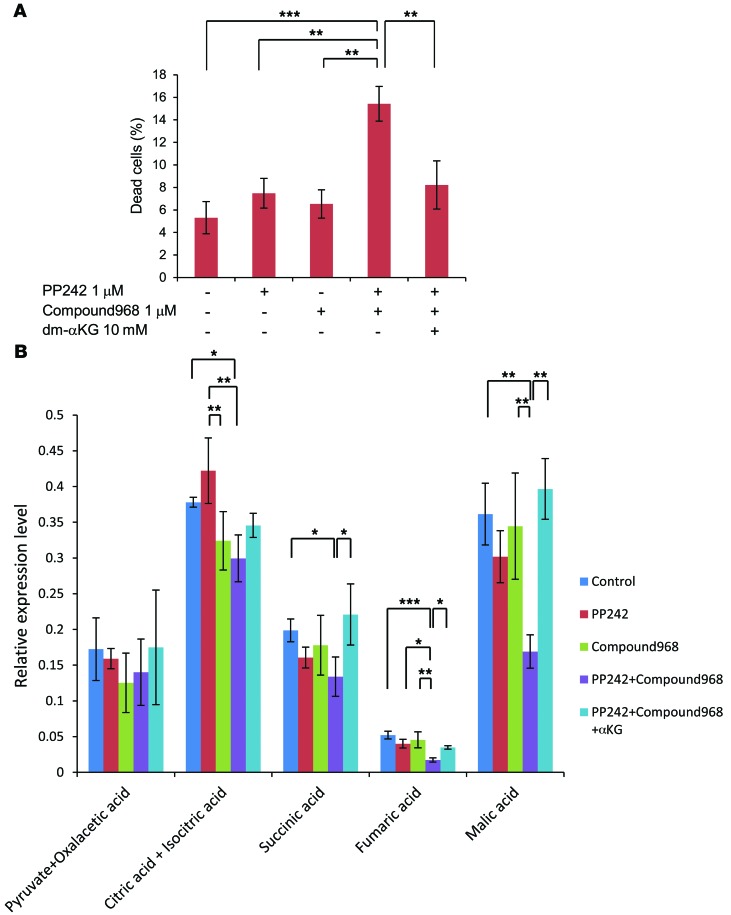
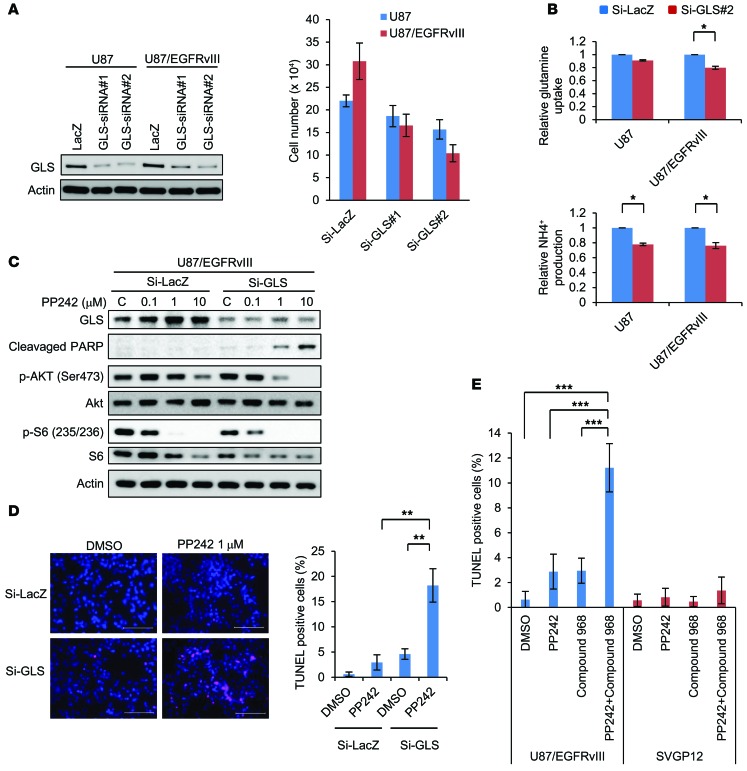
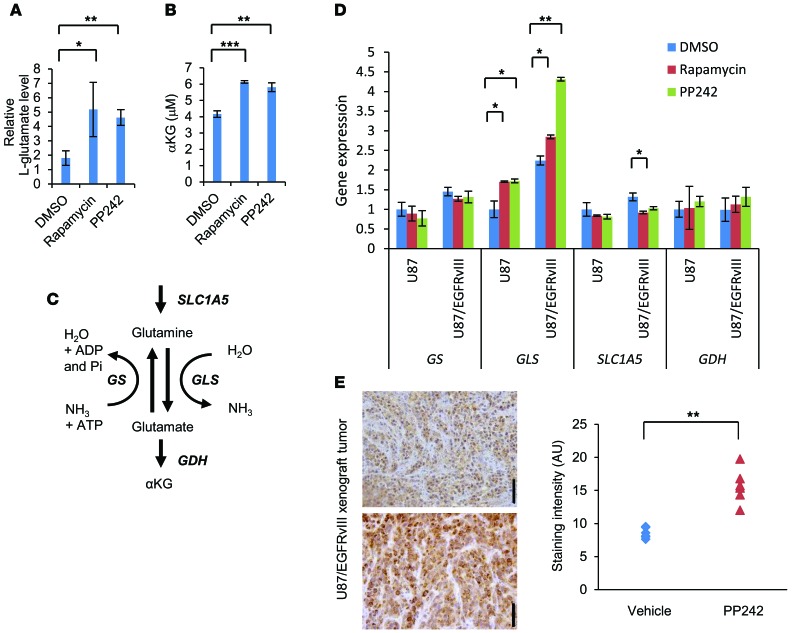
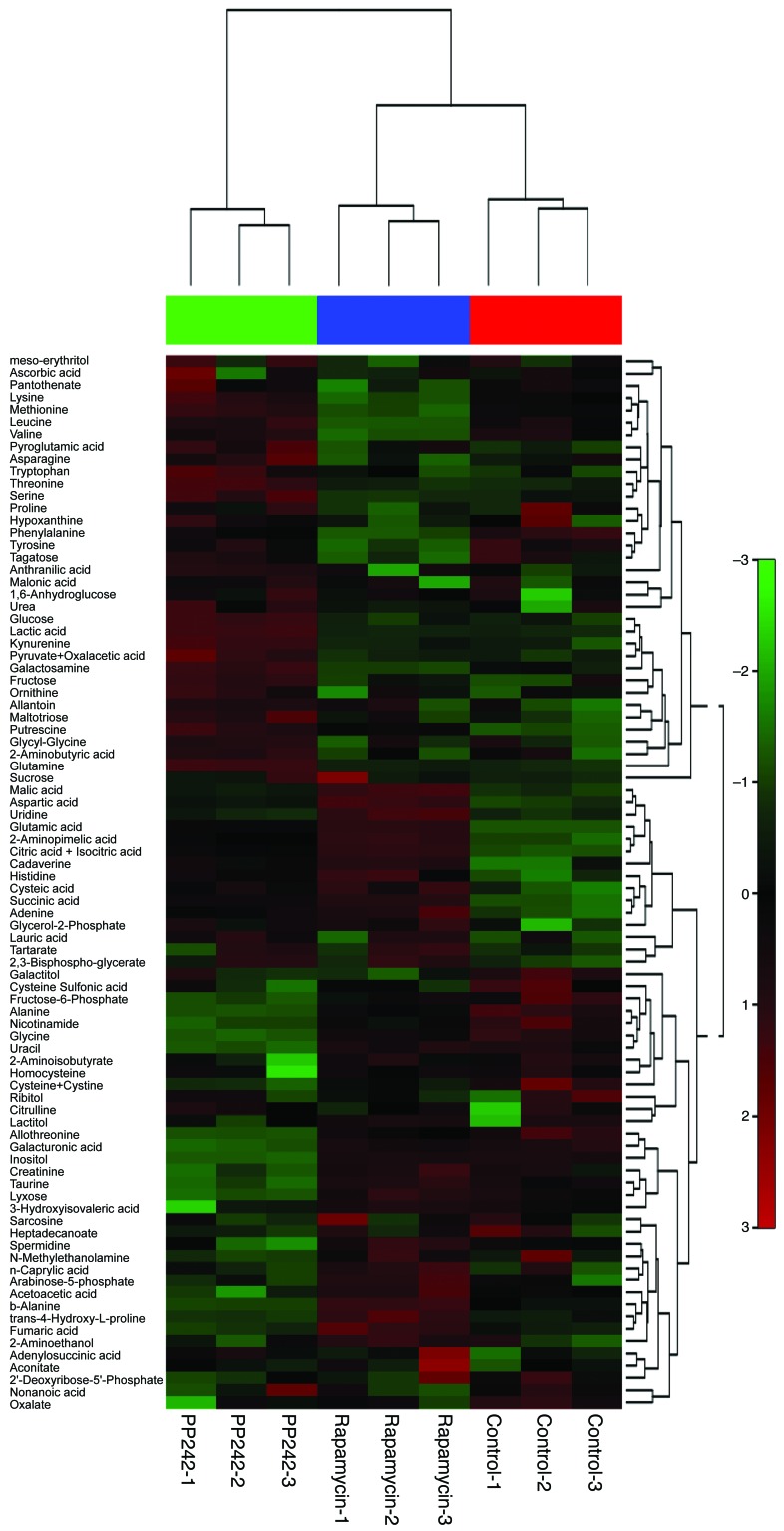
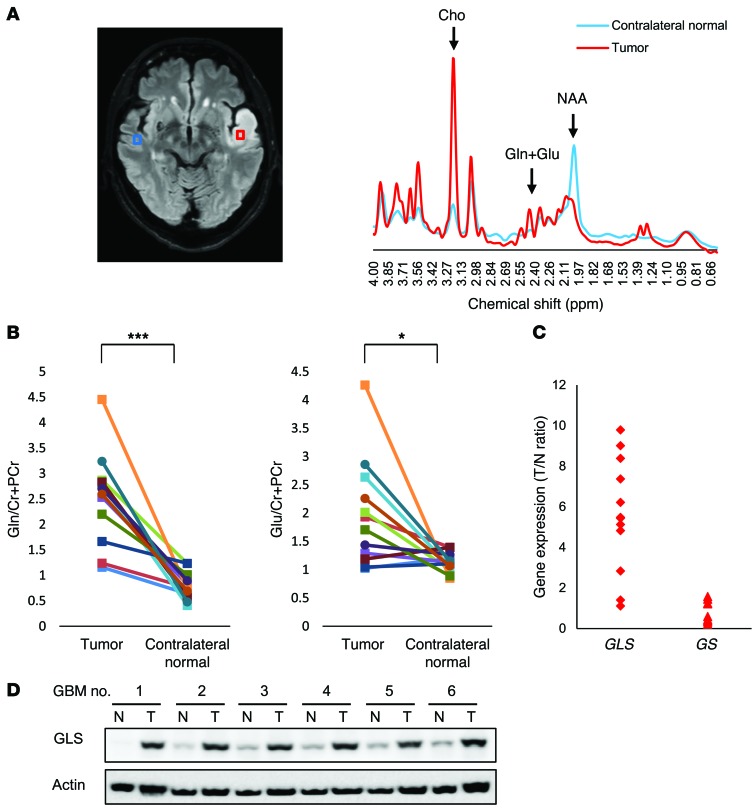
The mechanistic target of rapamycin (mTOR) is hyperactivated in many types of cancer, rendering it a compelling drug target; however, the impact of mTOR inhibition on metabolic reprogramming in cancer is incompletely understood. Here, by integrating metabolic and functional studies in glioblastoma multiforme (GBM) cell lines, preclinical models, and clinical samples, we demonstrate that the compensatory upregulation of glutamine metabolism promotes resistance to mTOR kinase inhibitors. Metabolomic studies in GBM cells revealed that glutaminase (GLS) and glutamate levels are elevated following mTOR kinase inhibitor treatment. Moreover, these mTOR inhibitor-dependent metabolic alterations were confirmed in a GBM xenograft model. Expression of GLS following mTOR inhibitor treatment promoted GBM survival in an α-ketoglutarate-dependent (αKG-dependent) manner. Combined genetic and/or pharmacological inhibition of mTOR kinase and GLS resulted in massive synergistic tumor cell death and growth inhibition in tumor-bearing mice. These results highlight a critical role for compensatory glutamine metabolism in promoting mTOR inhibitor resistance and suggest that rational combination therapy has the potential to suppress resistance.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
