

NOTCH reprograms mitochondrial metabolism for proinflammatory macrophage activation
- PMID: 25798621
- PMCID: PMC4396469
- DOI: 10.1172/JCI76468
NOTCH reprograms mitochondrial metabolism for proinflammatory macrophage activation
Abstract
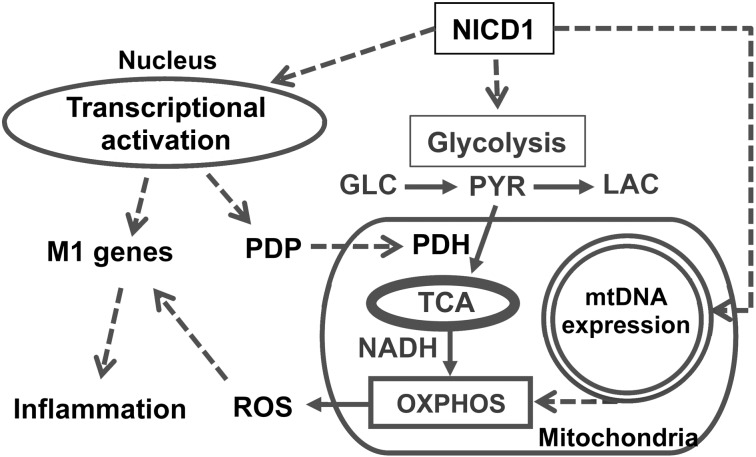
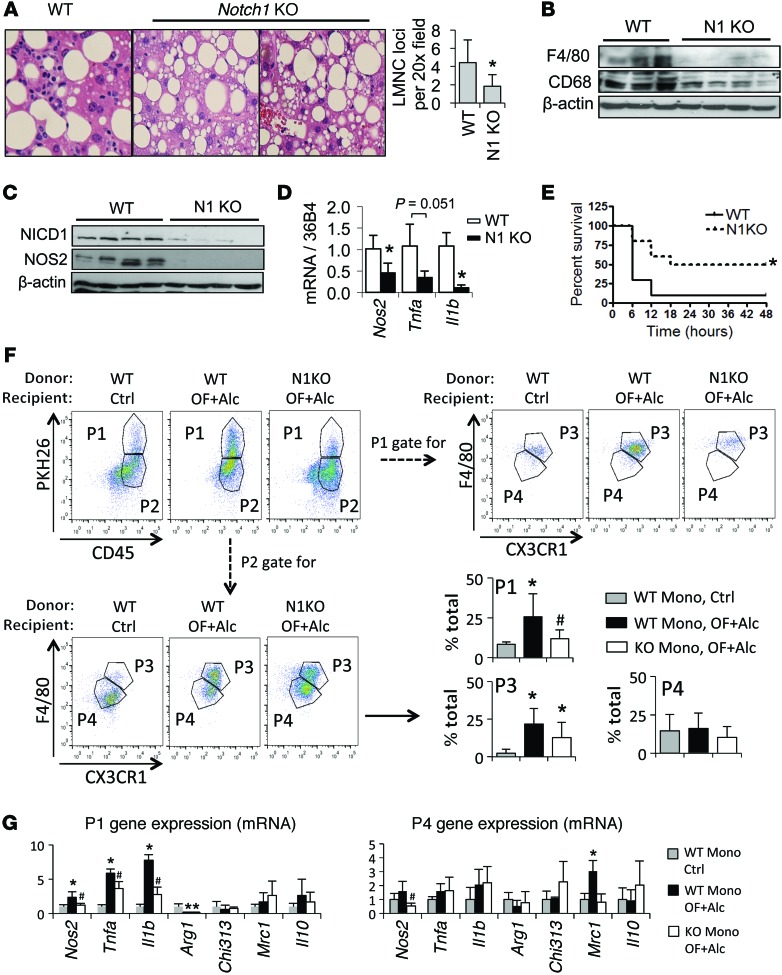
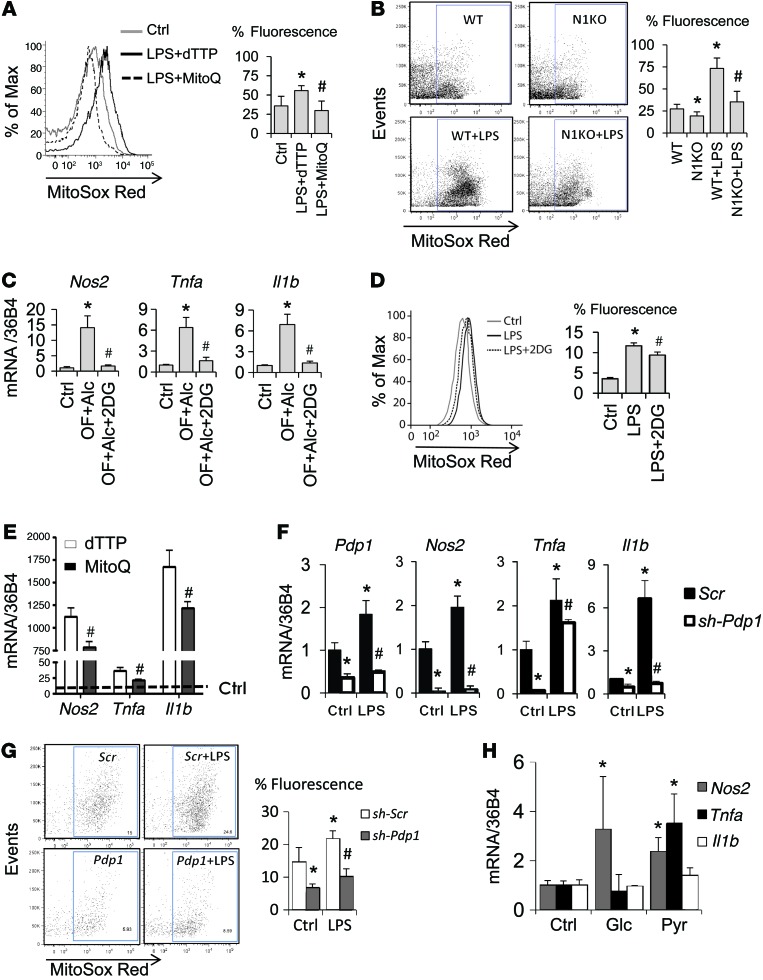
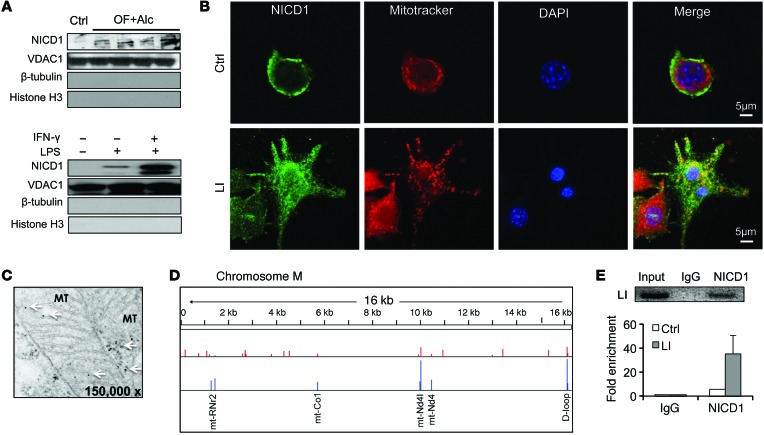
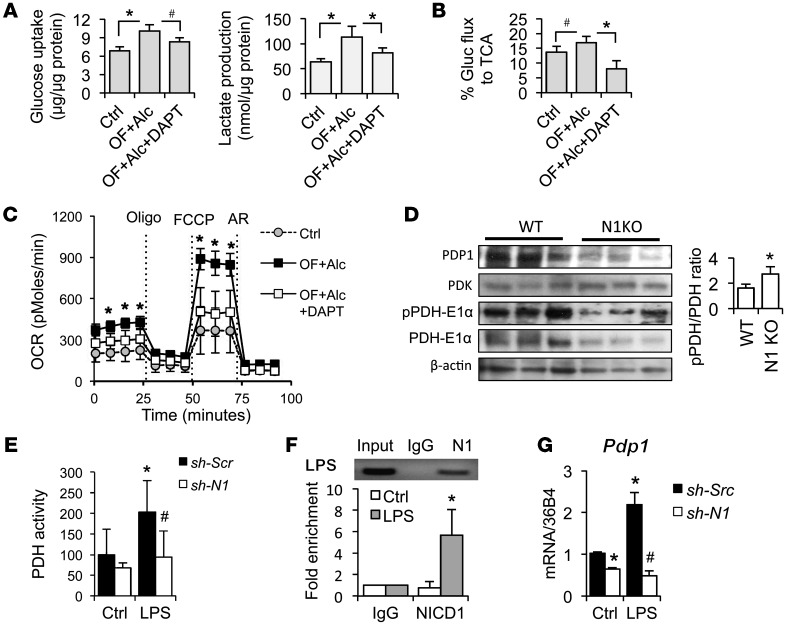
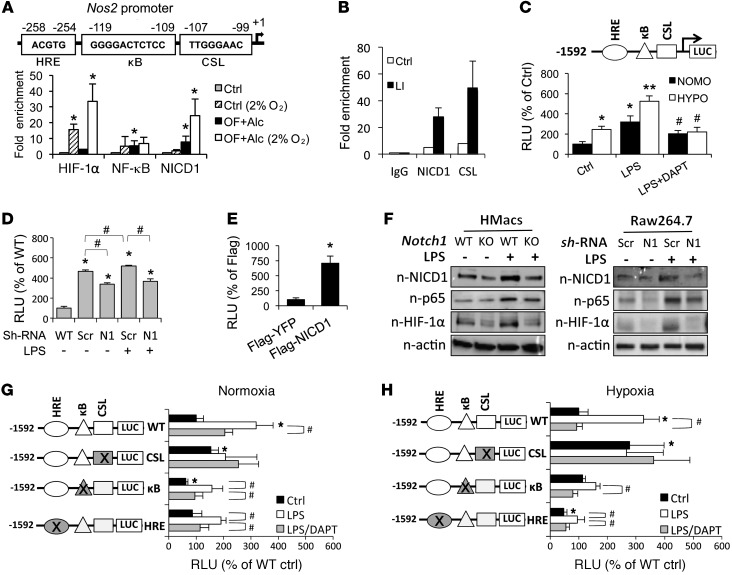
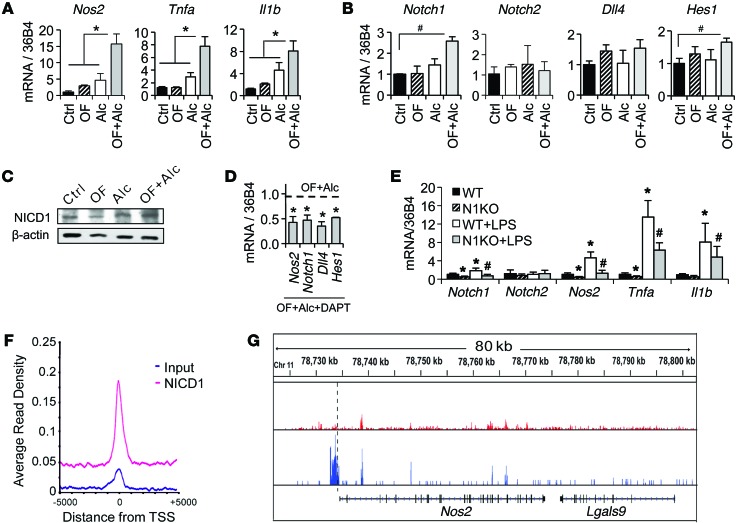
Metabolic reprogramming is implicated in macrophage activation, but the underlying mechanisms are poorly understood. Here, we demonstrate that the NOTCH1 pathway dictates activation of M1 phenotypes in isolated mouse hepatic macrophages (HMacs) and in a murine macrophage cell line by coupling transcriptional upregulation of M1 genes with metabolic upregulation of mitochondrial oxidative phosphorylation and ROS (mtROS) to augment induction of M1 genes. Enhanced mitochondrial glucose oxidation was achieved by increased recruitment of the NOTCH1 intracellular domain (NICD1) to nuclear and mitochondrial genes that encode respiratory chain components and by NOTCH-dependent induction of pyruvate dehydrogenase phosphatase 1 (Pdp1) expression, pyruvate dehydrogenase activity, and glucose flux to the TCA cycle. As such, inhibition of the NOTCH pathway or Pdp1 knockdown abrogated glucose oxidation, mtROS, and M1 gene expression. Conditional NOTCH1 deficiency in the myeloid lineage attenuated HMac M1 activation and inflammation in a murine model of alcoholic steatohepatitis and markedly reduced lethality following endotoxin-mediated fulminant hepatitis in mice. In vivo monocyte tracking further demonstrated the requirement of NOTCH1 for the migration of blood monocytes into the liver and subsequent M1 differentiation. Together, these results reveal that NOTCH1 promotes reprogramming of mitochondrial metabolism for M1 macrophage activation.
Figures
Comment in
-
Getting a NOTCH-up on Macrophage Activation in Alcoholic Liver Disease.Gastroenterology. 2015 Dec;149(7):1979-80. doi: 10.1053/j.gastro.2015.10.012. Epub 2015 Oct 20. Gastroenterology. 2015. PMID: 26496468 No abstract available.
-
Dual Notch signaling in proinflammatory macrophage activation.Hepatology. 2016 Apr;63(4):1381-3. doi: 10.1002/hep.28386. Epub 2016 Jan 21. Hepatology. 2016. PMID: 26662111 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
