

Mutations in HPCA cause autosomal-recessive primary isolated dystonia
- PMID: 25799108
- PMCID: PMC4385177
- DOI: 10.1016/j.ajhg.2015.02.007
Mutations in HPCA cause autosomal-recessive primary isolated dystonia
Abstract
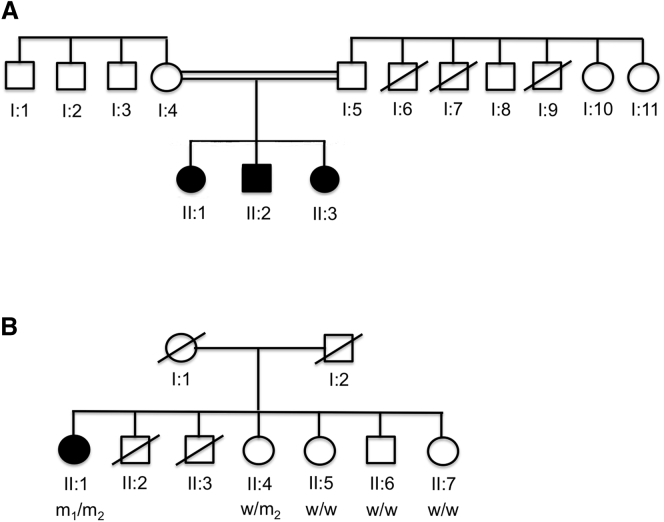
Reports of primary isolated dystonia inherited in an autosomal-recessive (AR) manner, often lumped together as "DYT2 dystonia," have appeared in the scientific literature for several decades, but no genetic cause has been identified to date. Using a combination of homozygosity mapping and whole-exome sequencing in a consanguineous kindred affected by AR isolated dystonia, we identified homozygous mutations in HPCA, a gene encoding a neuronal calcium sensor protein found almost exclusively in the brain and at particularly high levels in the striatum, as the cause of disease in this family. Subsequently, compound-heterozygous mutations in HPCA were also identified in a second independent kindred affected by AR isolated dystonia. Functional studies suggest that hippocalcin might play a role in regulating voltage-dependent calcium channels. The identification of mutations in HPCA as a cause of AR primary isolated dystonia paves the way for further studies to assess whether "DYT2 dystonia" is a genetically homogeneous condition or not.
Copyright © 2015 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures


Comment in
-
Mutations in hippocalcin and autosomal recessive dystonia: a role for perturbed calcium signaling?Mov Disord. 2015 Jun;30(7):911. doi: 10.1002/mds.26261. Epub 2015 May 23. Mov Disord. 2015. PMID: 26009984 No abstract available.
-
DYT2 revealed: Hippocalcin mutations cause autosomal-recessive isolated dystonia.Mov Disord. 2015 Nov;30(13):1725. doi: 10.1002/mds.26280. Epub 2015 Jun 12. Mov Disord. 2015. PMID: 26094611 No abstract available.
-
Mystery surrounding DYT2 dystonia now solved: HPCA mutations identified in DYT2-like family.Mov Disord. 2015 Jul;30(8):1035. doi: 10.1002/mds.26288. Epub 2015 Jun 22. Mov Disord. 2015. PMID: 26095160 No abstract available.
References
-
- Fahn S. Concept and classification of dystonia. Adv. Neurol. 1988;50:1–8. - PubMed
-
- Ozelius L.J., Hewett J.W., Page C.E., Bressman S.B., Kramer P.L., Shalish C., de Leon D., Brin M.F., Raymond D., Corey D.P. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat. Genet. 1997;17:40–48. - PubMed
-
- Fuchs T., Gavarini S., Saunders-Pullman R., Raymond D., Ehrlich M.E., Bressman S.B., Ozelius L.J. Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat. Genet. 2009;41:286–288. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
