

REV7 counteracts DNA double-strand break resection and affects PARP inhibition
- PMID: 25799992
- PMCID: PMC4671316
- DOI: 10.1038/nature14328
REV7 counteracts DNA double-strand break resection and affects PARP inhibition
Abstract
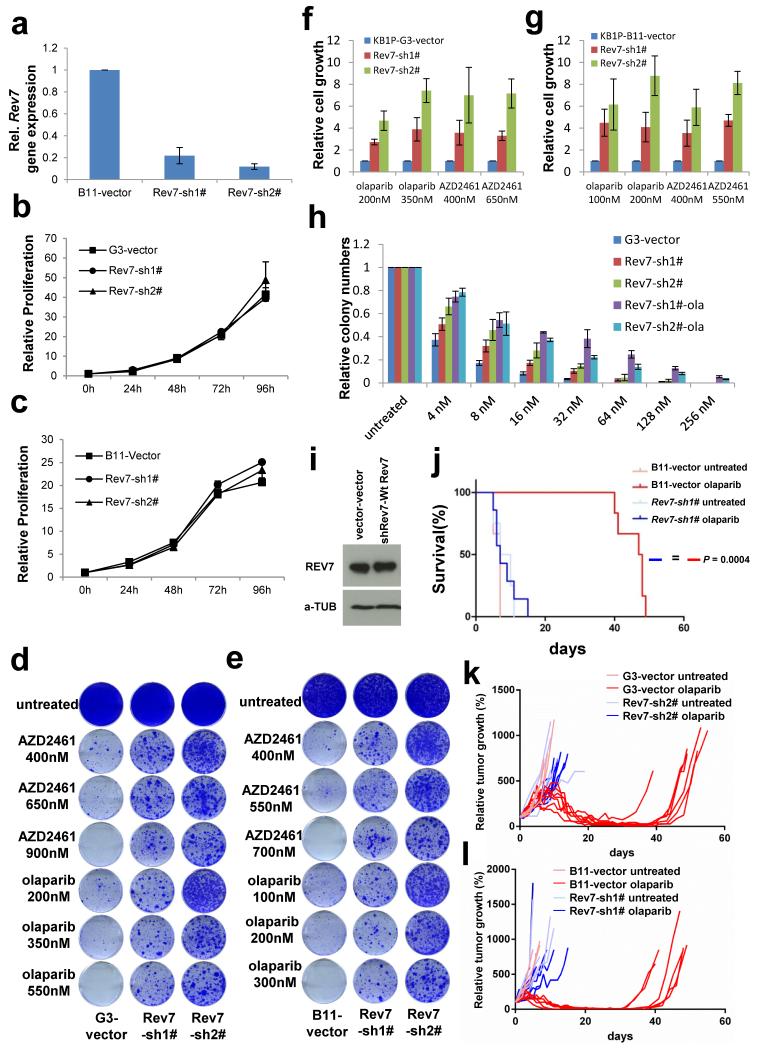
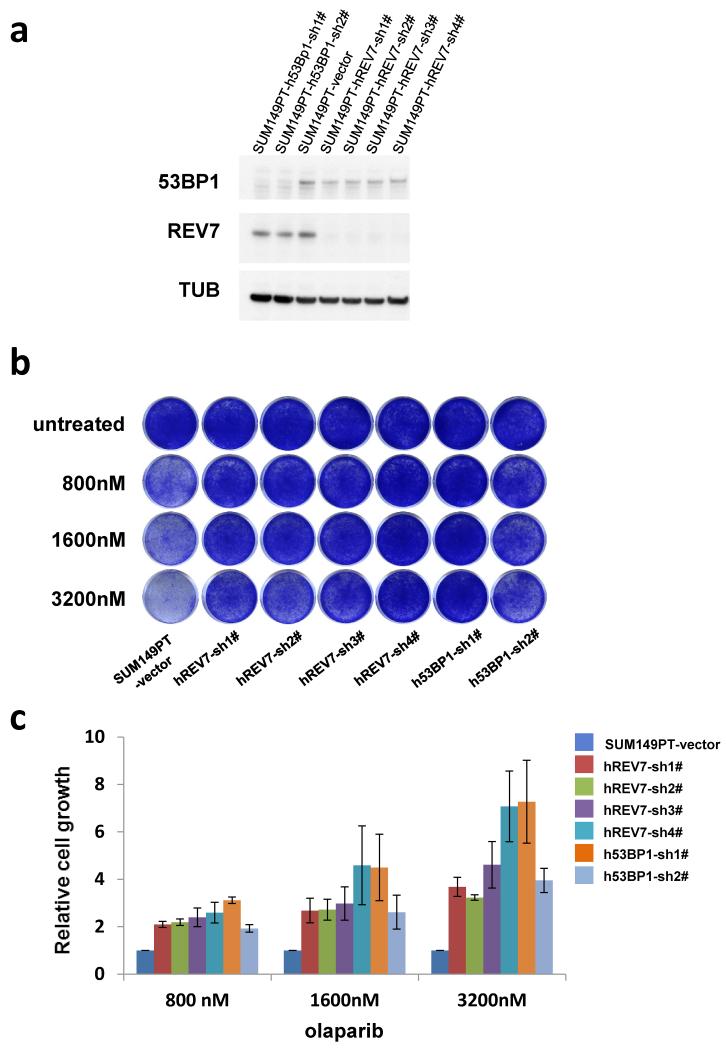
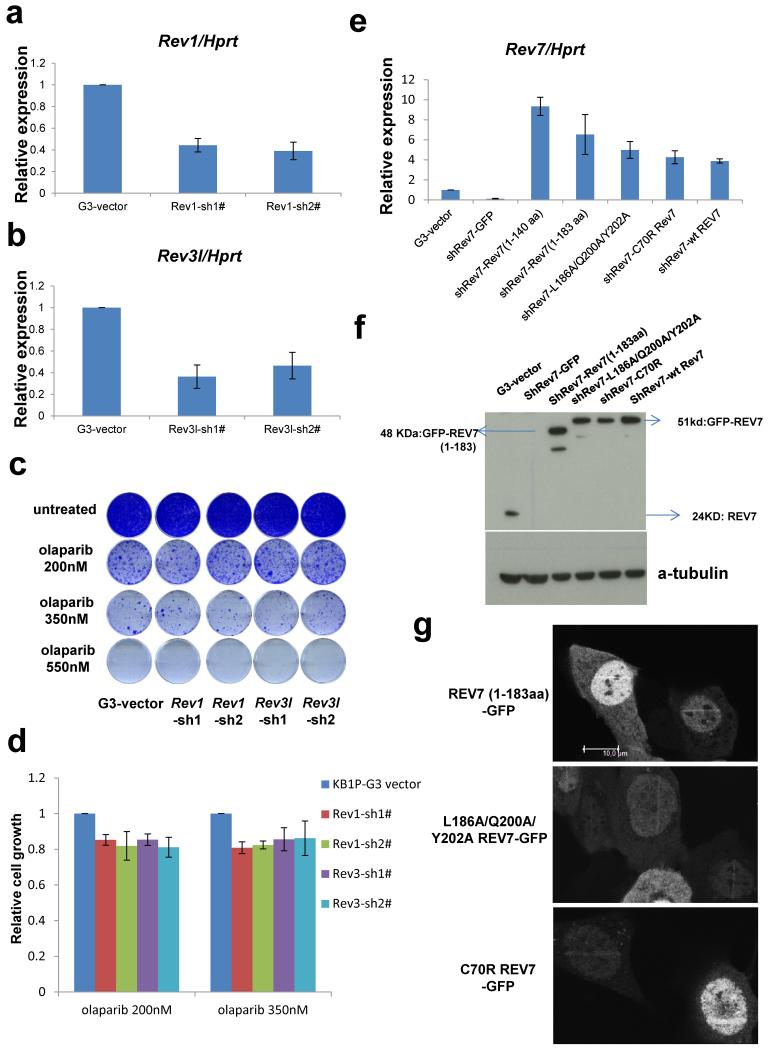
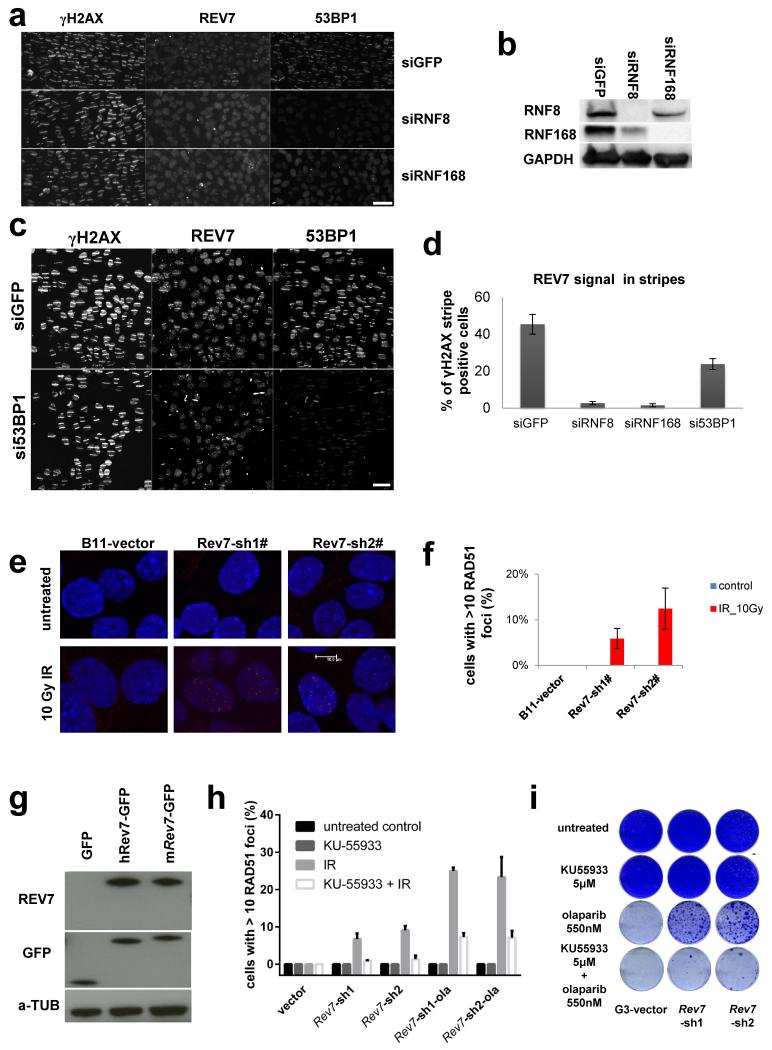
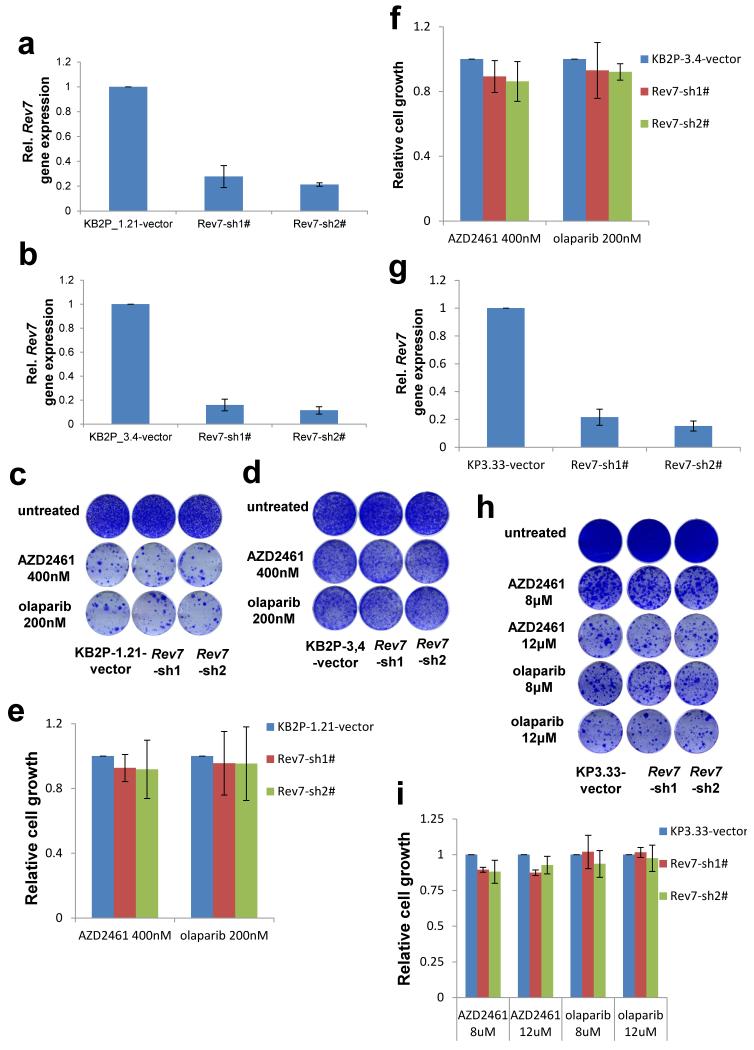
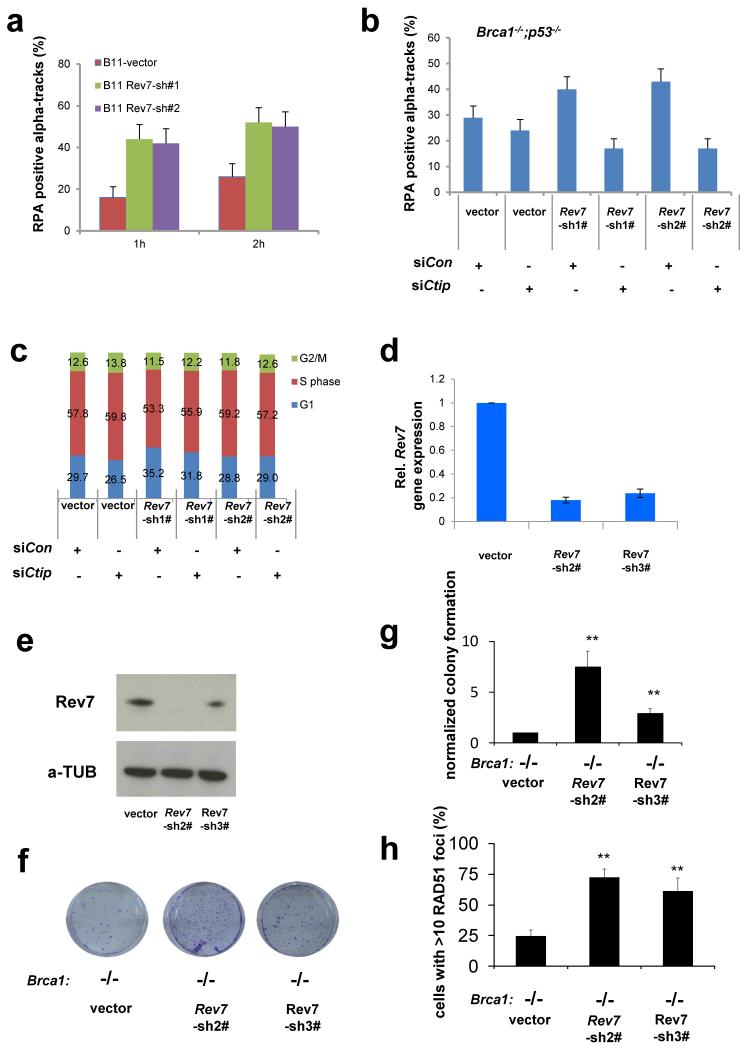
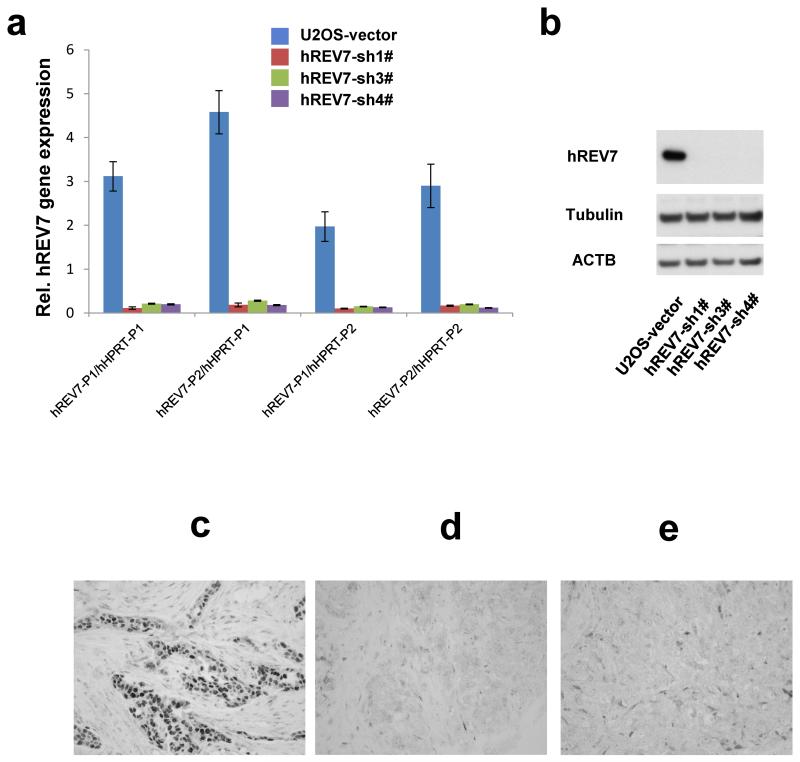
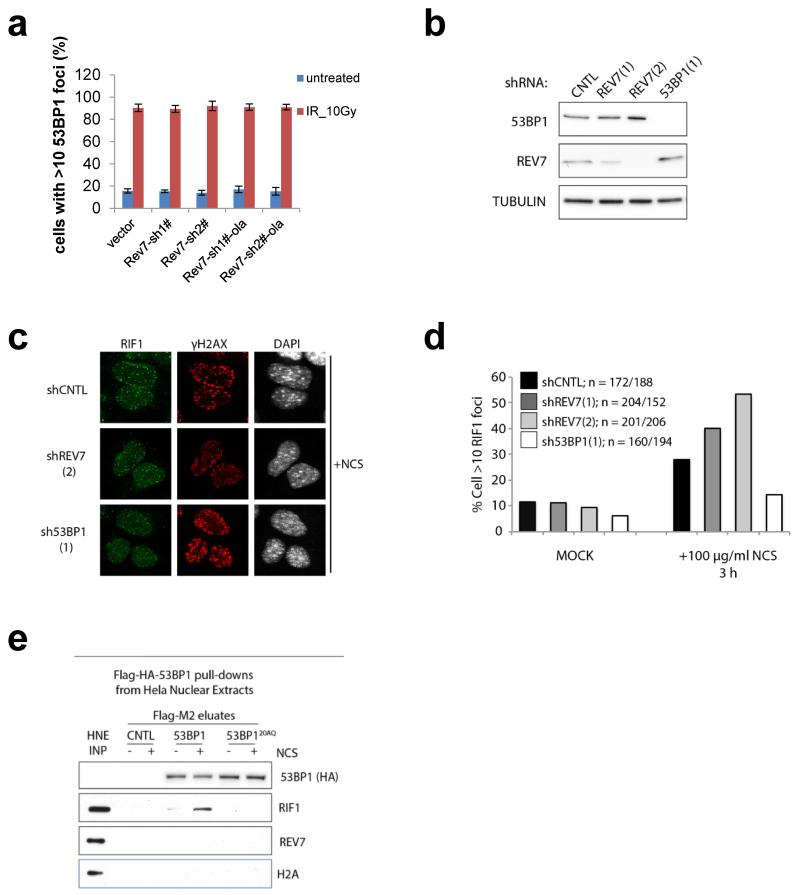
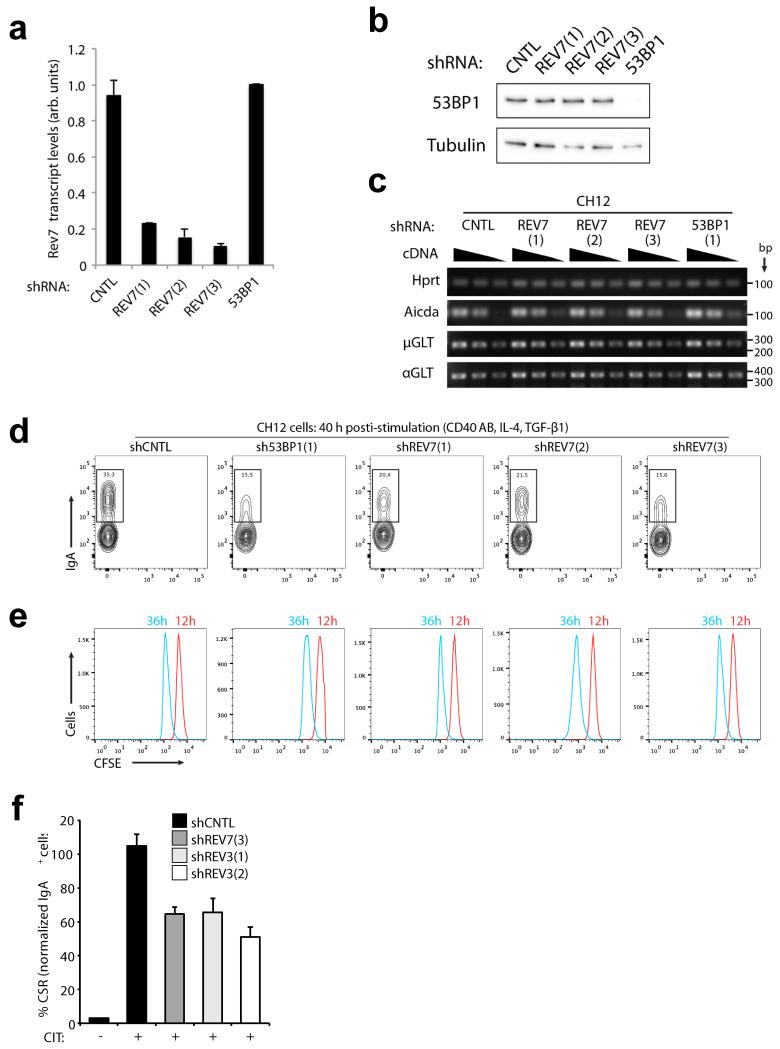
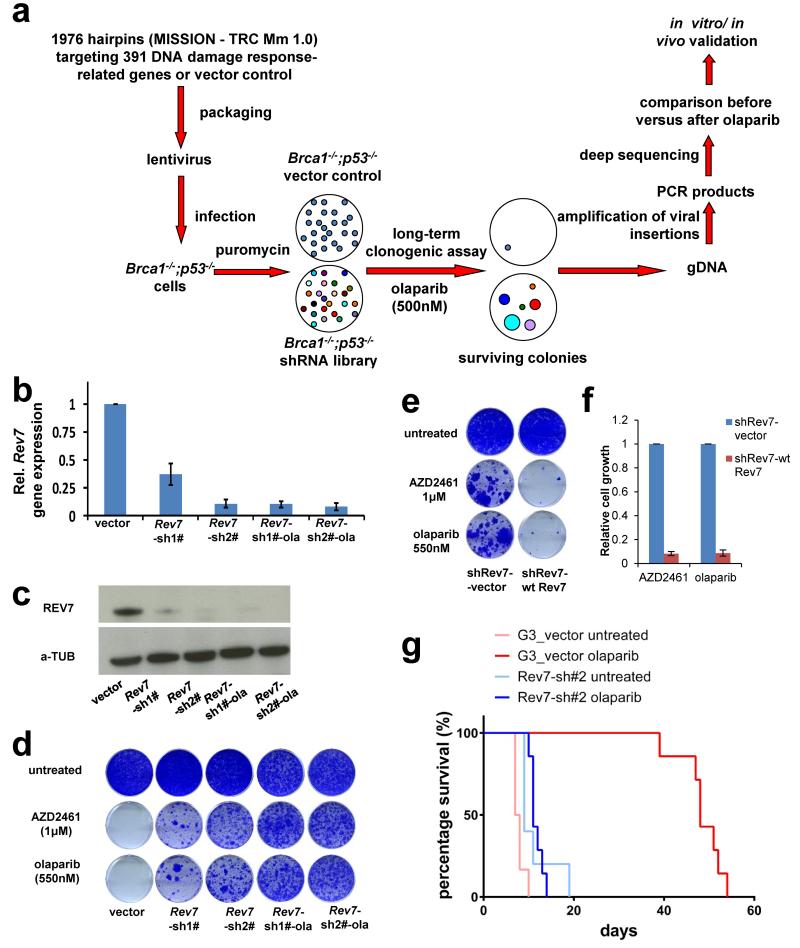
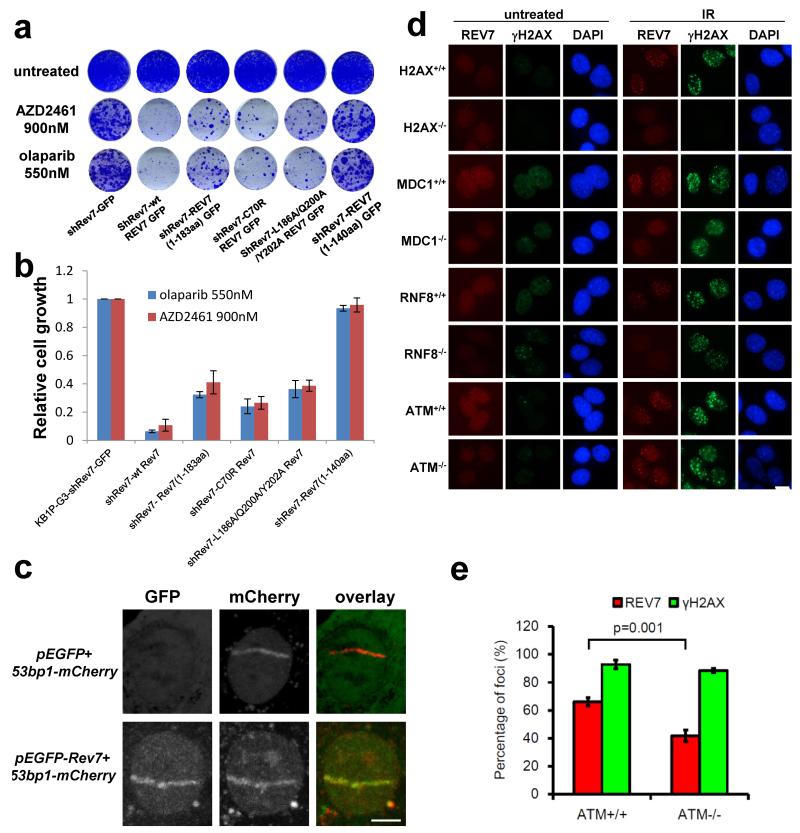
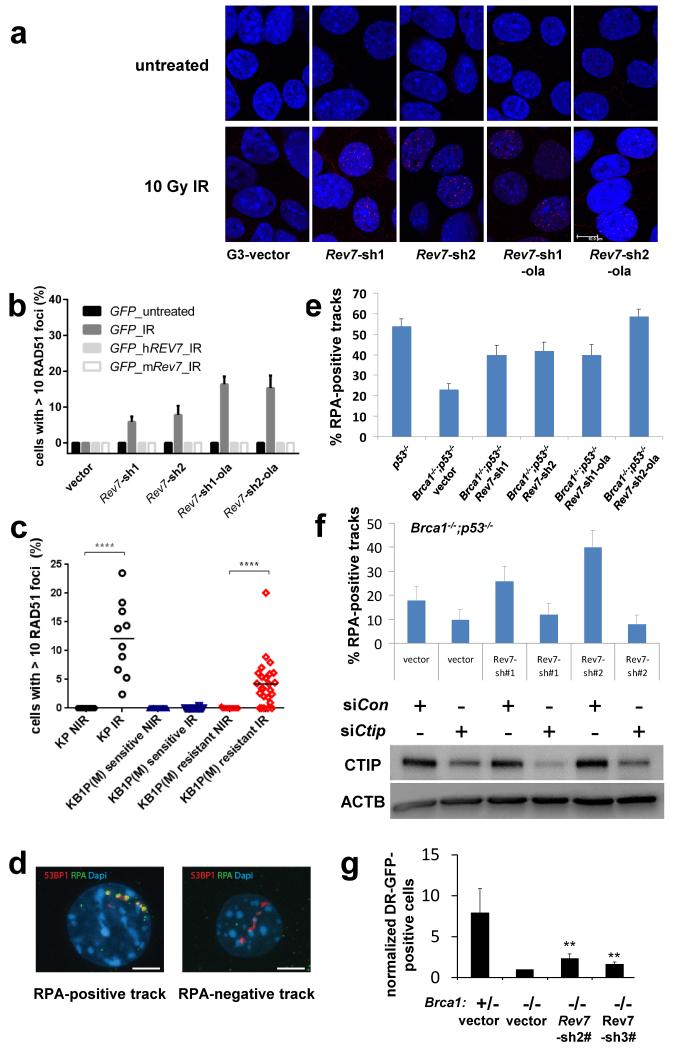
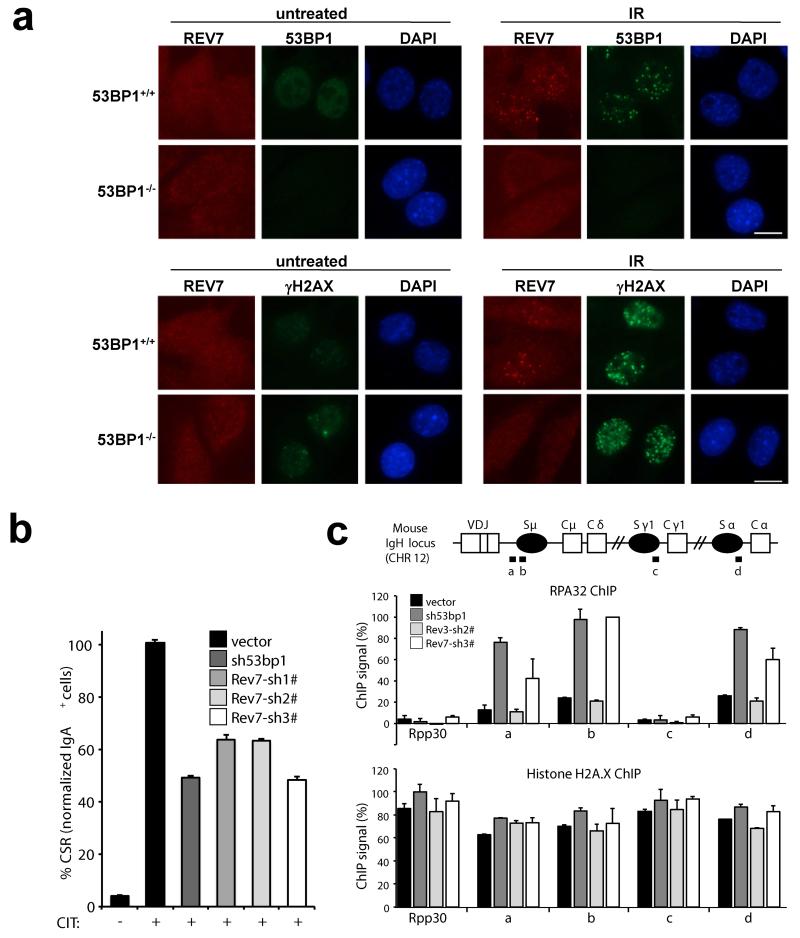
Error-free repair of DNA double-strand breaks (DSBs) is achieved by homologous recombination (HR), and BRCA1 is an important factor for this repair pathway. In the absence of BRCA1-mediated HR, the administration of PARP inhibitors induces synthetic lethality of tumour cells of patients with breast or ovarian cancers. Despite the benefit of this tailored therapy, drug resistance can occur by HR restoration. Genetic reversion of BRCA1-inactivating mutations can be the underlying mechanism of drug resistance, but this does not explain resistance in all cases. In particular, little is known about BRCA1-independent restoration of HR. Here we show that loss of REV7 (also known as MAD2L2) in mouse and human cell lines re-establishes CTIP-dependent end resection of DSBs in BRCA1-deficient cells, leading to HR restoration and PARP inhibitor resistance, which is reversed by ATM kinase inhibition. REV7 is recruited to DSBs in a manner dependent on the H2AX-MDC1-RNF8-RNF168-53BP1 chromatin pathway, and seems to block HR and promote end joining in addition to its regulatory role in DNA damage tolerance. Finally, we establish that REV7 blocks DSB resection to promote non-homologous end-joining during immunoglobulin class switch recombination. Our results reveal an unexpected crucial function of REV7 downstream of 53BP1 in coordinating pathological DSB repair pathway choices in BRCA1-deficient cells.
Figures
Comment in
-
REV7/MAD2L2: the multitasking maestro emerges as a barrier to recombination.EMBO J. 2015 Jun 12;34(12):1609-11. doi: 10.15252/embj.201591697. Epub 2015 Apr 20. EMBO J. 2015. PMID: 25896508 Free PMC article.
References
-
- Farmer H, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. - PubMed
-
- Lord CJ, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat. Med. 2013;19:1381–1388. - PubMed
-
- Ang JE, et al. Efficacy of chemotherapy in BRCA1/2 mutation carrier ovarian cancer in the setting of PARP inhibitor resistance: a multi-institutional study. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013;19:5485–5493. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
