

Microglial M1/M2 polarization and metabolic states
- PMID: 25800044
- PMCID: PMC4742299
- DOI: 10.1111/bph.13139
Microglial M1/M2 polarization and metabolic states
Abstract
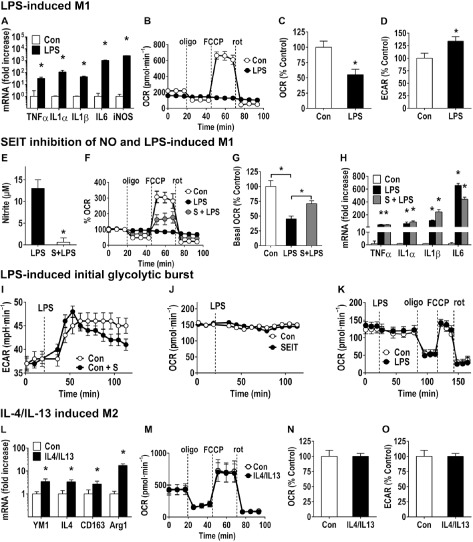
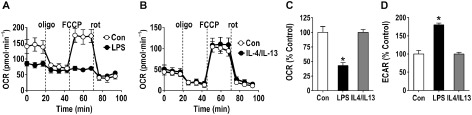
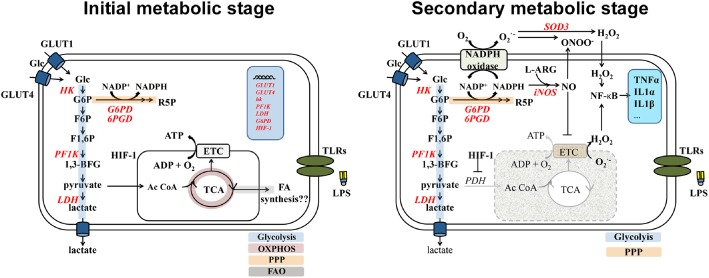
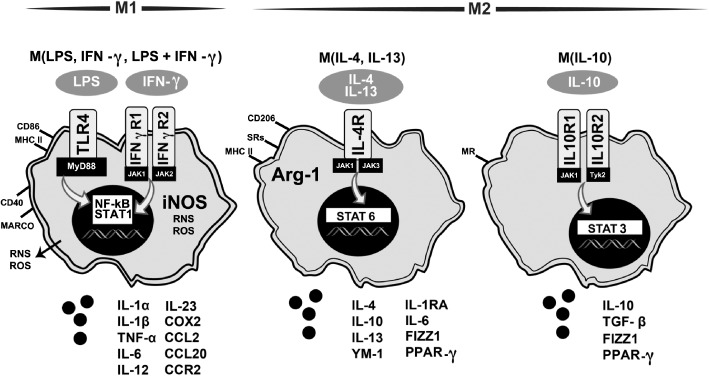
Microglia are critical nervous system-specific immune cells serving as tissue-resident macrophages influencing brain development, maintenance of the neural environment, response to injury and repair. As influenced by their environment, microglia assume a diversity of phenotypes and retain the capability to shift functions to maintain tissue homeostasis. In comparison with peripheral macrophages, microglia demonstrate similar and unique features with regards to phenotype polarization, allowing for innate immunological functions. Microglia can be stimulated by LPS or IFN-γ to an M1 phenotype for expression of pro-inflammatory cytokines or by IL-4/IL-13 to an M2 phenotype for resolution of inflammation and tissue repair. Increasing evidence suggests a role of metabolic reprogramming in the regulation of the innate inflammatory response. Studies using peripheral immune cells demonstrate that polarization to an M1 phenotype is often accompanied by a shift in cells from oxidative phosphorylation to aerobic glycolysis for energy production. More recently, the link between polarization and mitochondrial energy metabolism has been considered in microglia. Under these conditions, energy demands would be associated with functional activities and cell survival and thus, may serve to influence the contribution of microglia activation to various neurodegenerative conditions. This review examines the polarization states of microglia and their relationship to mitochondrial metabolism. Additional supporting experimental data are provided to demonstrate mitochondrial metabolic shifts in primary microglia and the BV-2 microglia cell line induced under LPS (M1) and IL-4/IL-13 (M2) polarization.
Published 2015. This article is a U.S. Government work and is in the public domain in the USA.
Figures
References
-
- Alliot F, Godin I, Pessac B (1999). Microglia derived from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res 117: 145–152. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
