

Activation of necroptosis in multiple sclerosis
- PMID: 25801023
- PMCID: PMC4494996
- DOI: 10.1016/j.celrep.2015.02.051
Activation of necroptosis in multiple sclerosis
Abstract
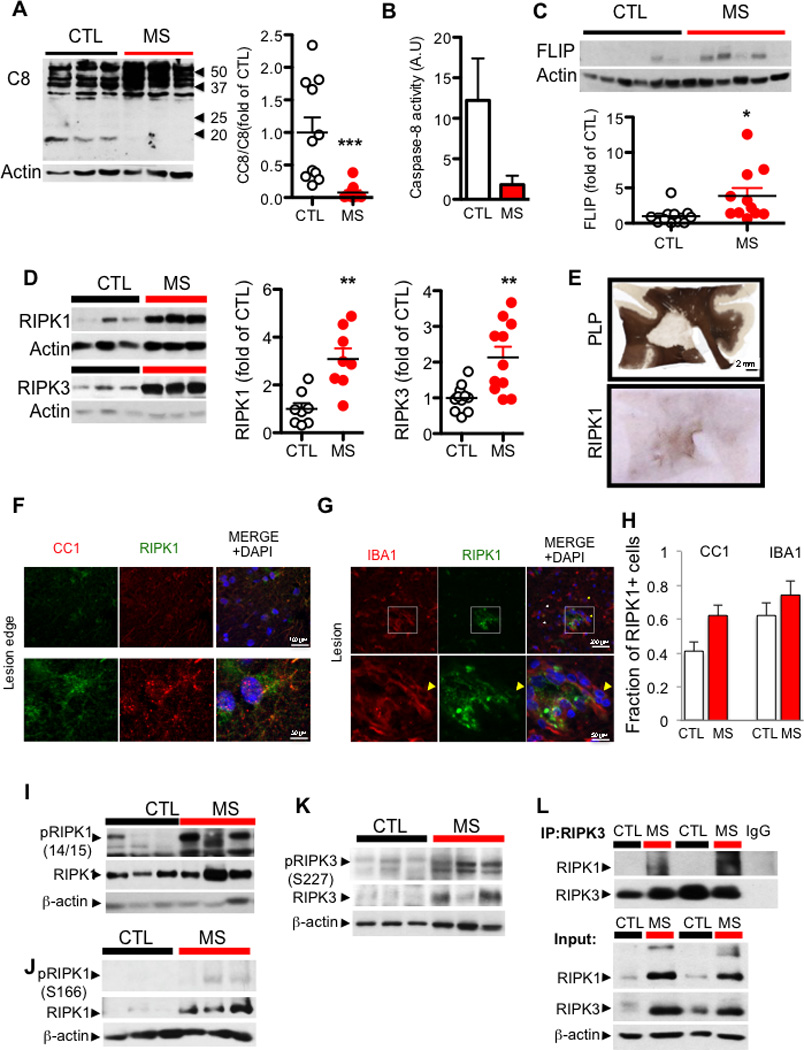
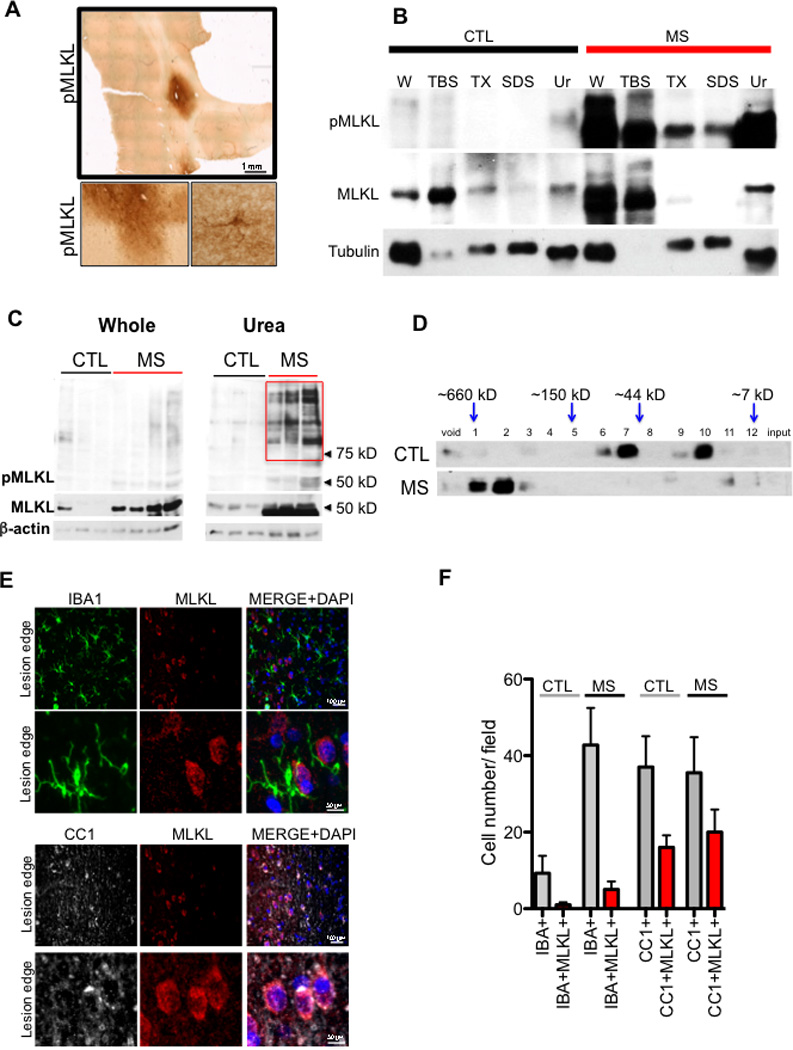
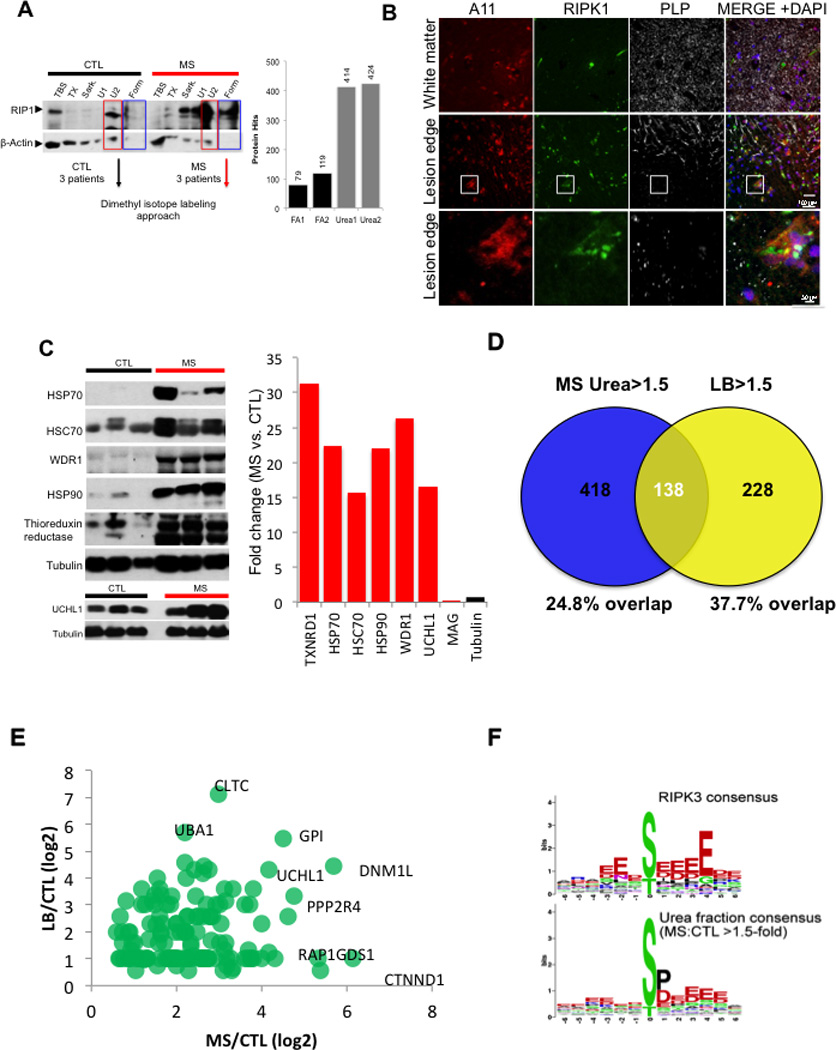
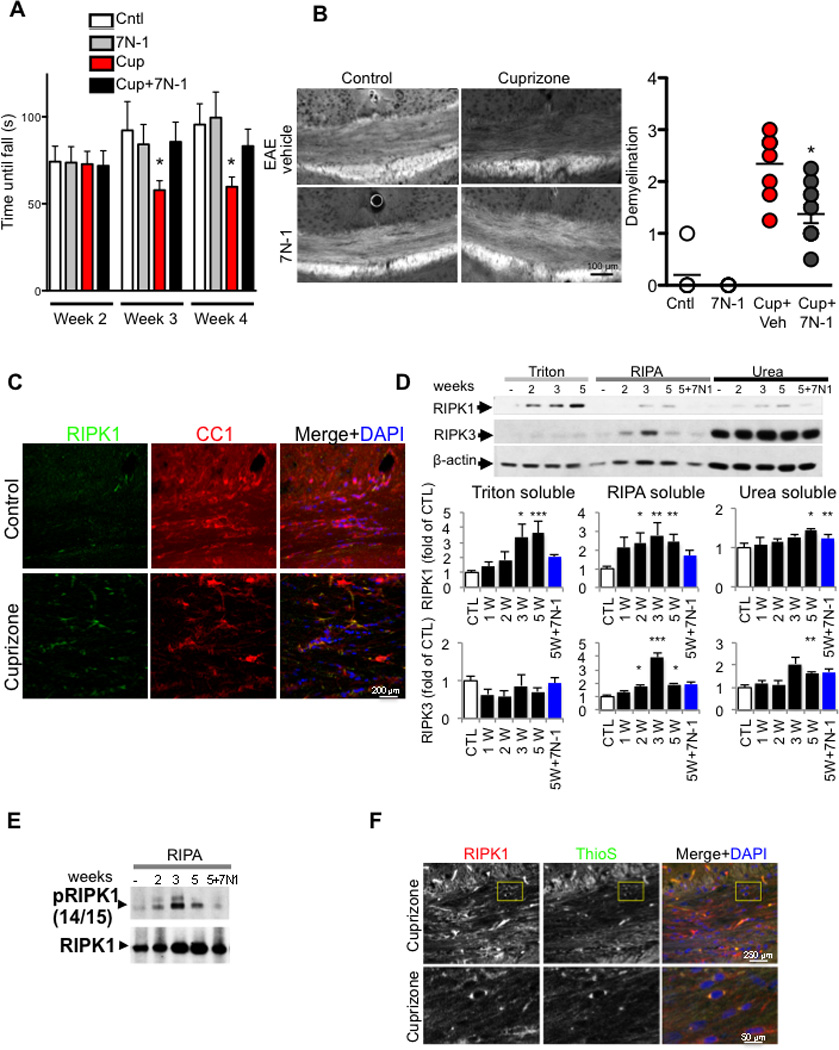
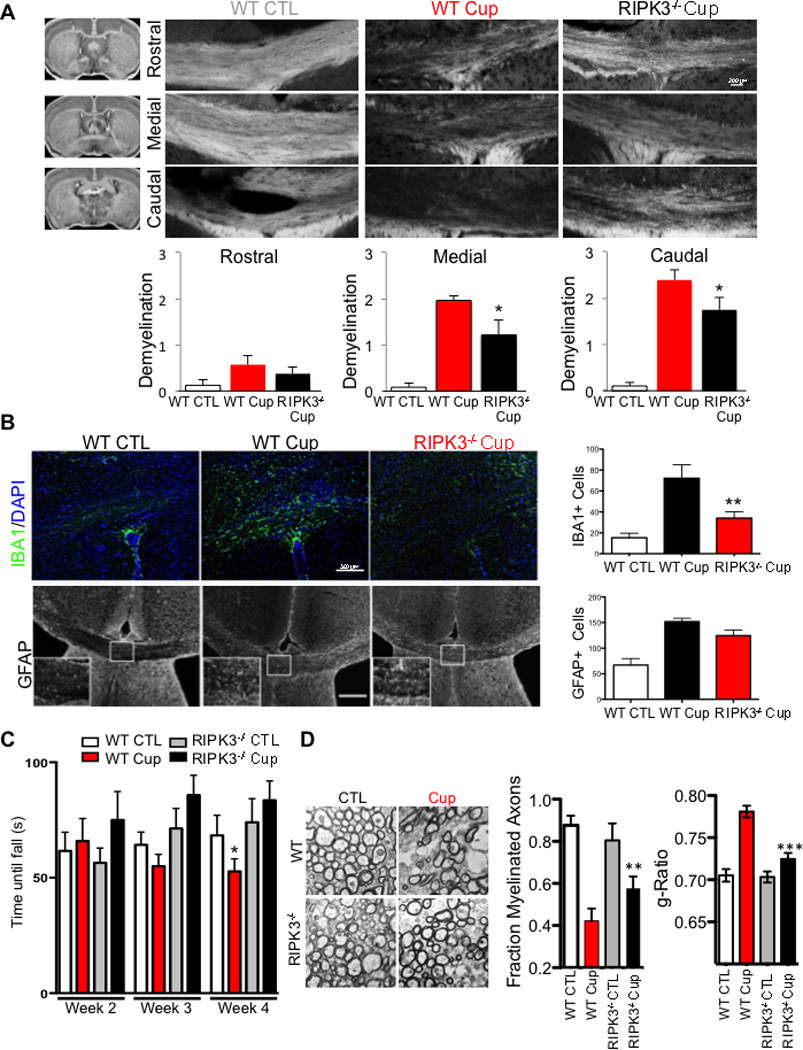
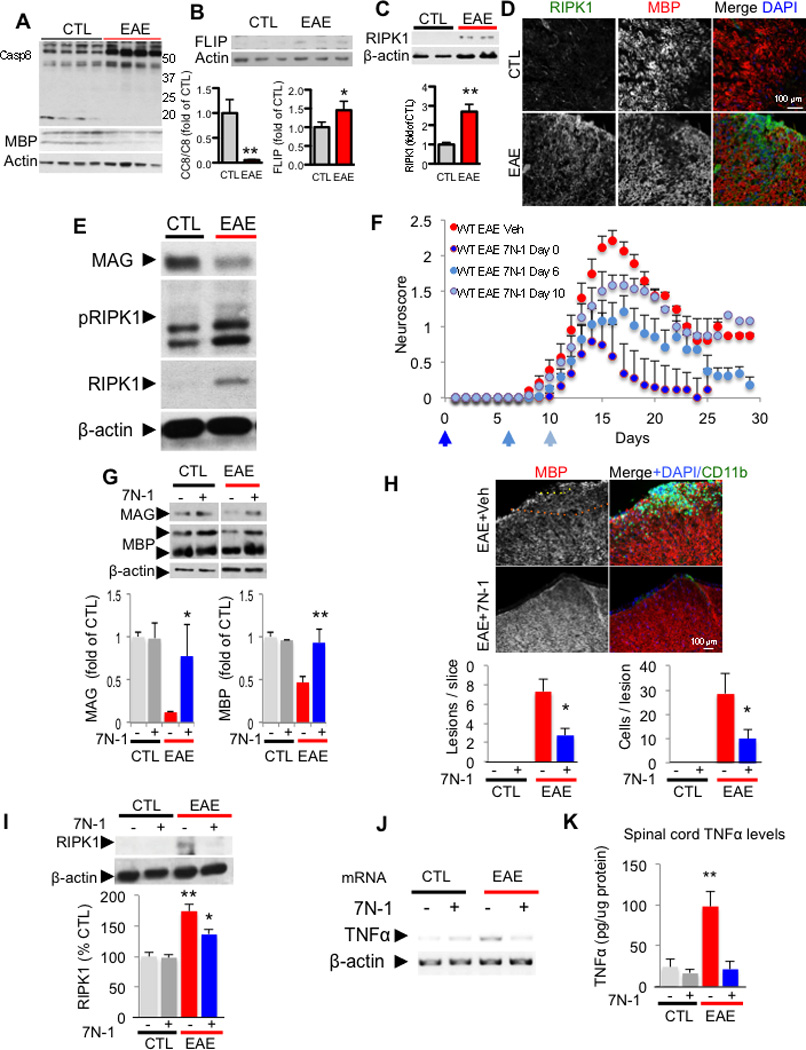
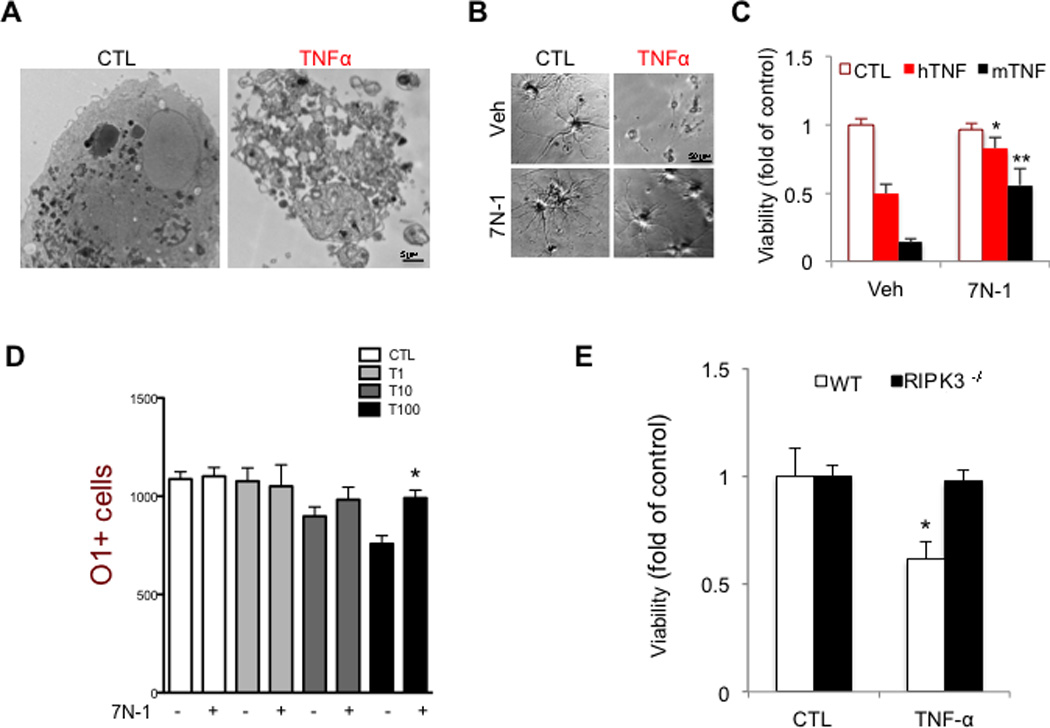
Multiple sclerosis (MS), a common neurodegenerative disease of the CNS, is characterized by the loss of oligodendrocytes and demyelination. Tumor necrosis factor α (TNF-α), a proinflammatory cytokine implicated in MS, can activate necroptosis, a necrotic cell death pathway regulated by RIPK1 and RIPK3 under caspase-8-deficient conditions. Here, we demonstrate defective caspase-8 activation, as well as activation of RIPK1, RIPK3, and MLKL, the hallmark mediators of necroptosis, in the cortical lesions of human MS pathological samples. Furthermore, we show that MS pathological samples are characterized by an increased insoluble proteome in common with other neurodegenerative diseases such as Alzheimer's disease (AD), Parkinson’s disease (PD), and Huntington's disease (HD). Finally, we show that necroptosis mediates oligodendrocyte degeneration induced by TNF-α and that inhibition of RIPK1 protects against oligodendrocyte cell death in two animal models of MS and in culture. Our findings demonstrate that necroptosis is involved in MS and suggest that targeting RIPK1 may represent a therapeutic strategy for MS.
Figures
References
-
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, et al. TDP-43 is a component of ubiquitin-positive taunegative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. - PubMed
-
- Arnett HA, Fancy SP, Alberta JA, Zhao C, Plant SR, Kaing S, Raine CS, Rowitch DH, Franklin RJ, Stiles CD. bHLH transcription factor Olig1 is required to repair demyelinated lesions in the CNS. Science. 2004;306:2111–2115. - PubMed
-
- Berger SB, Kasparcova V, Hoffman S, Swift B, Dare L, Schaeffer M, Capriotti C, Cook M, Finger J, Hughes-Earle A, et al. Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J Immunol. 2014;192:5476–5480. - PMC - PubMed
-
- Blakemore WF. Observations on oligodendrocyte degeneration, the resolution of status spongiosus and remyelination in cuprizone intoxication in mice. J Neurocytol. 1972;1:413–426. - PubMed
-
- Buck D, Hemmer B. Treatment of multiple sclerosis: current concepts and future perspectives. J Neurol. 2011;258:1747–1762. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
