

Origins and impacts of new mammalian exons
- PMID: 25801031
- PMCID: PMC4715653
- DOI: 10.1016/j.celrep.2015.02.058
Origins and impacts of new mammalian exons
Abstract
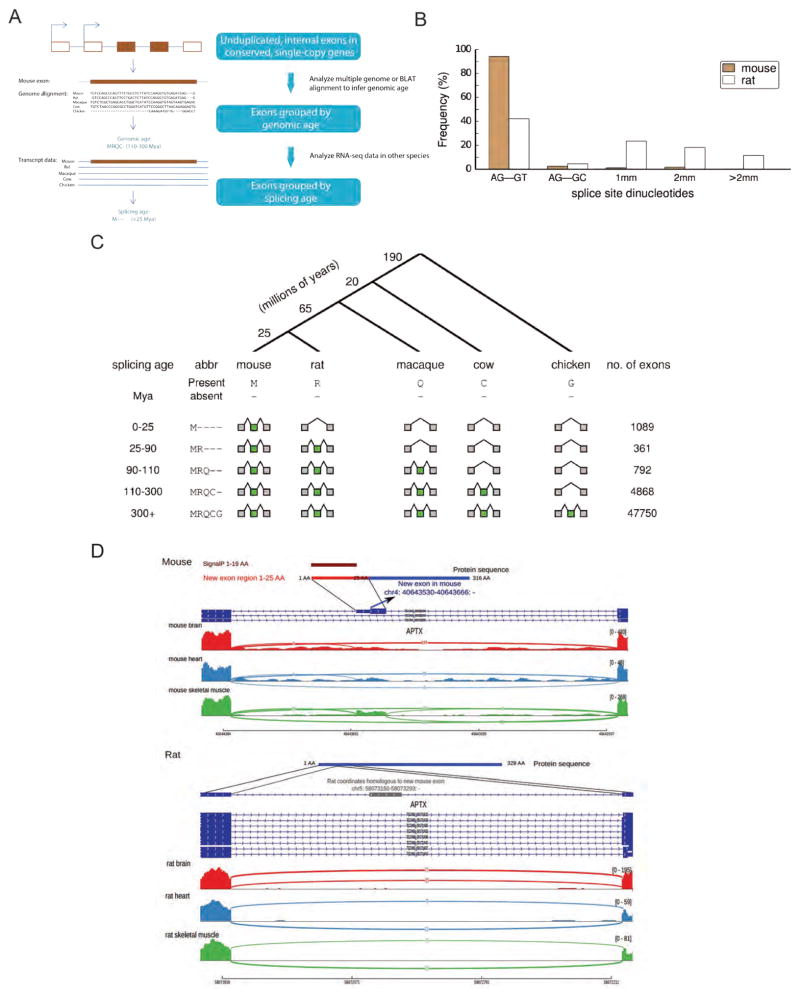
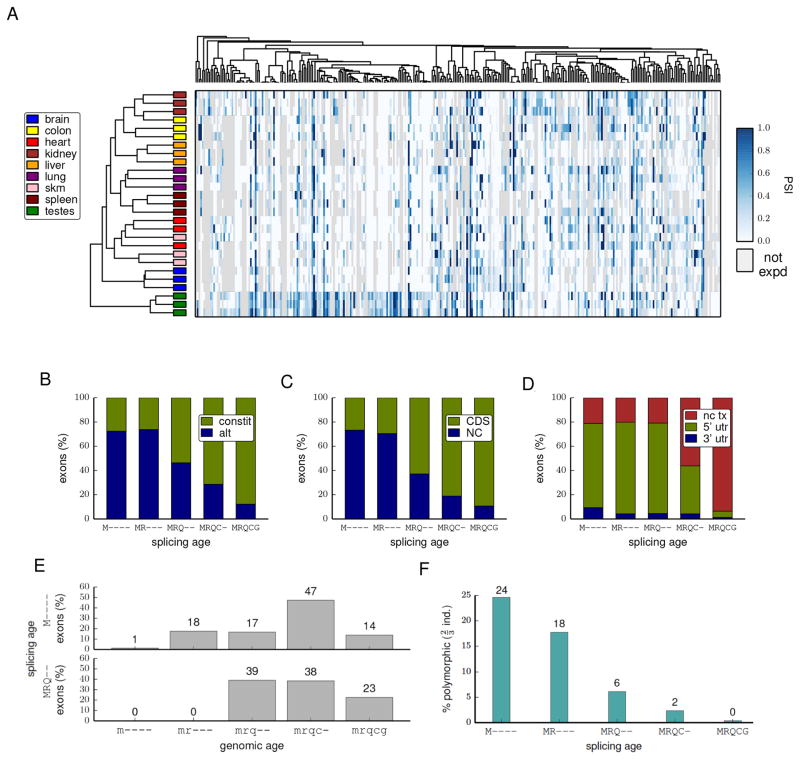
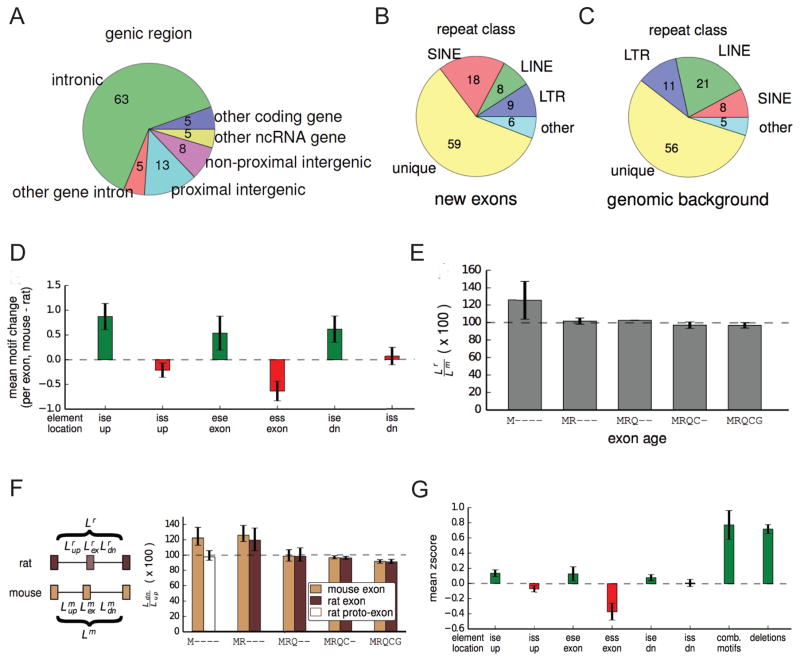
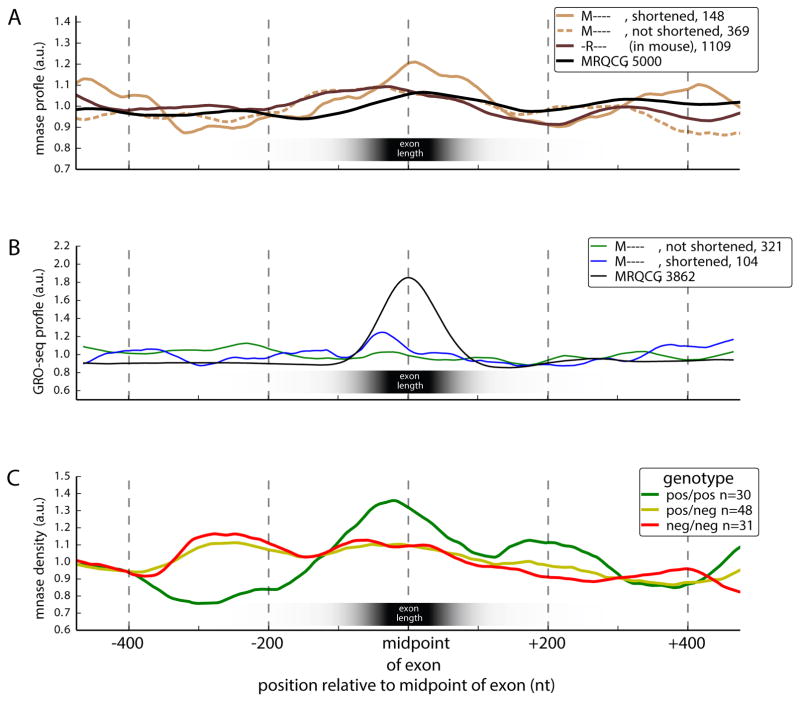
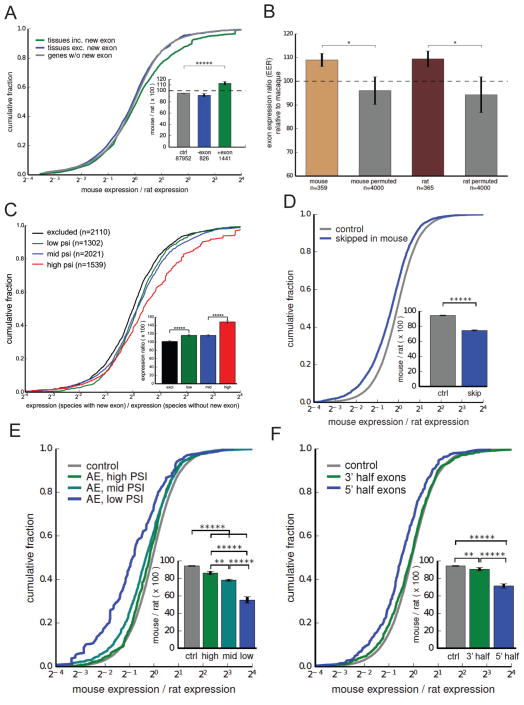
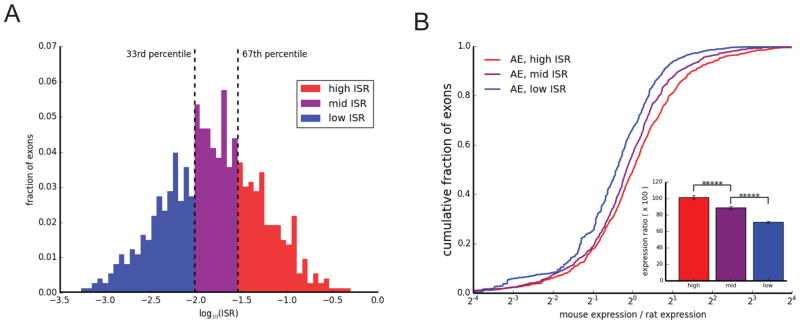
Mammalian genes are composed of exons, but the evolutionary origins and functions of new internal exons are poorly understood. Here, we analyzed patterns of exon gain using deep cDNA sequencing data from five mammals and one bird, identifying thousands of species- and lineage-specific exons. Most new exons derived from unique rather than repetitive intronic sequence. Unlike exons conserved across mammals, species-specific internal exons were mostly located in 5' UTRs and alternatively spliced. They were associated with upstream intronic deletions, increased nucleosome occupancy, and RNA polymerase II pausing. Genes containing new internal exons had increased gene expression, but only in tissues in which the exon was included. Increased expression correlated with the level of exon inclusion, promoter proximity, and signatures of cotranscriptional splicing. Altogether, these findings suggest that increased splicing at the 5' ends of genes enhances expression and that changes in 5' end splicing alter gene expression between tissues and between species.
Copyright © 2015 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- BARBOSA-MORAIS NL, IRIMIA M, PAN Q, XIONG HY, GUEROUSSOV S, LEE LJ, SLOBODENIUC V, KUTTER C, WATT S, COLAK R, KIM T, MISQUITTA-ALI CM, WILSON MD, KIM PM, ODOM DT, FREY BJ, BLENCOWE BJ. The evolutionary landscape of alternative splicing in vertebrate species. Science. 2012;338:1587–93. - PubMed
-
- BENDTSEN JD, NIELSEN H, VON HEIJNE G, BRUNAK S. Improved prediction of signal peptides: SignalP 3.0. Journal of molecular biology. 2004;340:783–95. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
