

High-resolution chromatin dynamics during a yeast stress response
- PMID: 25801168
- PMCID: PMC4405355
- DOI: 10.1016/j.molcel.2015.02.002
High-resolution chromatin dynamics during a yeast stress response
Abstract
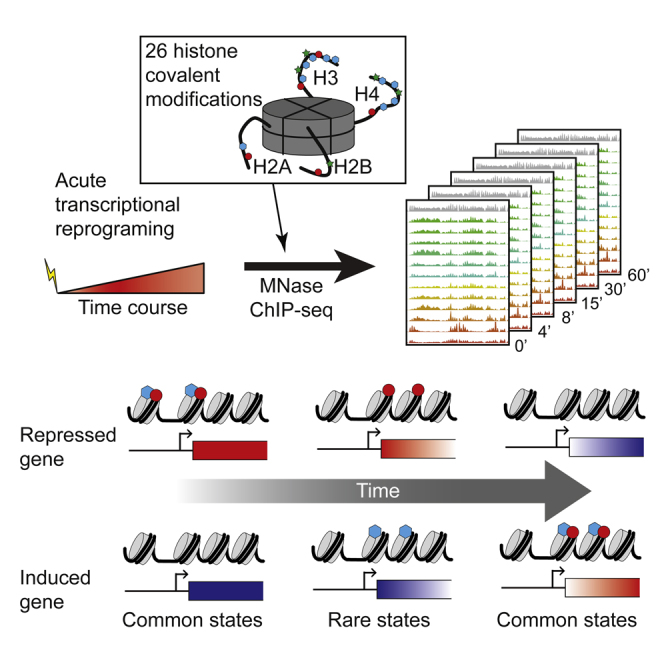
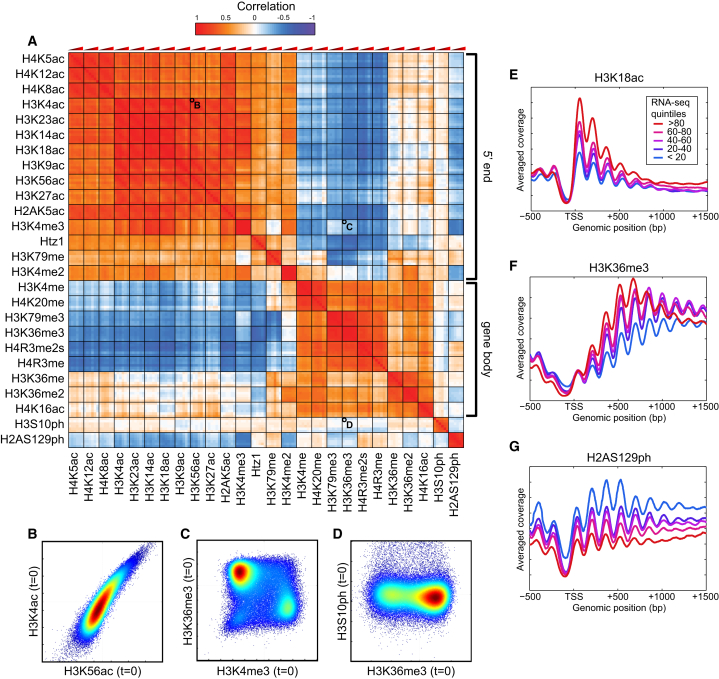
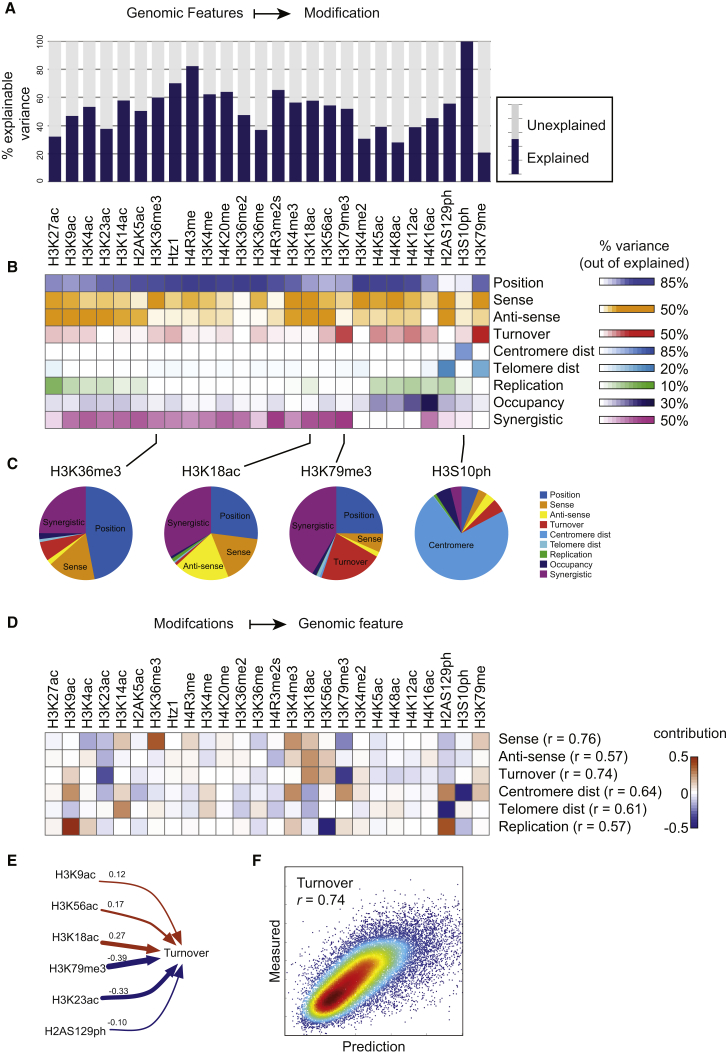
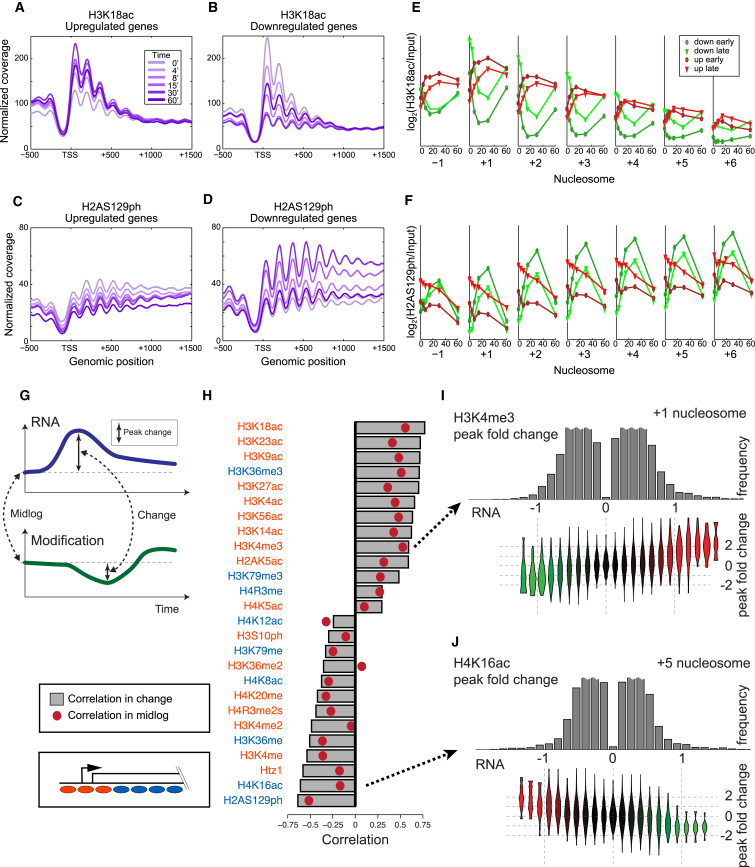
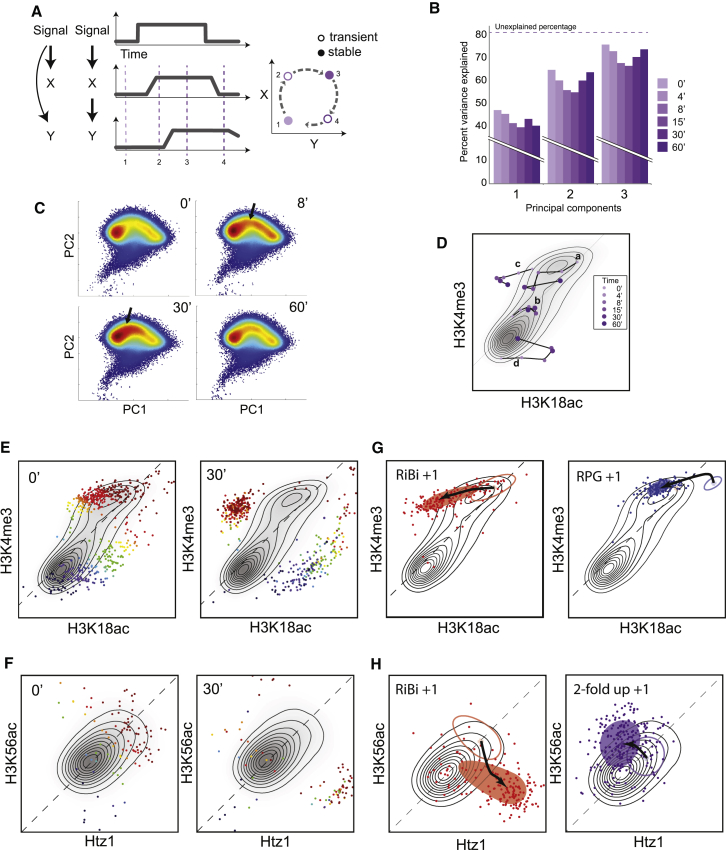
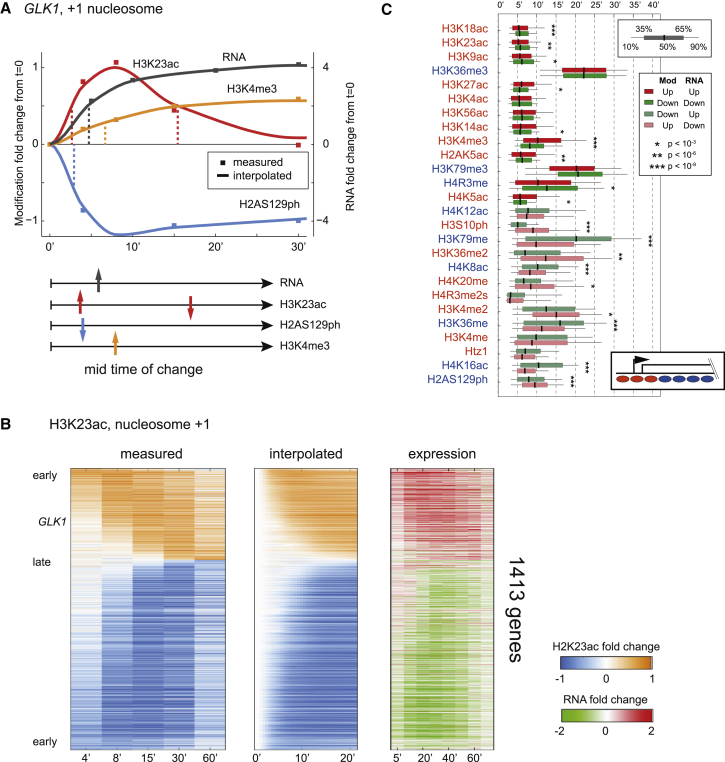
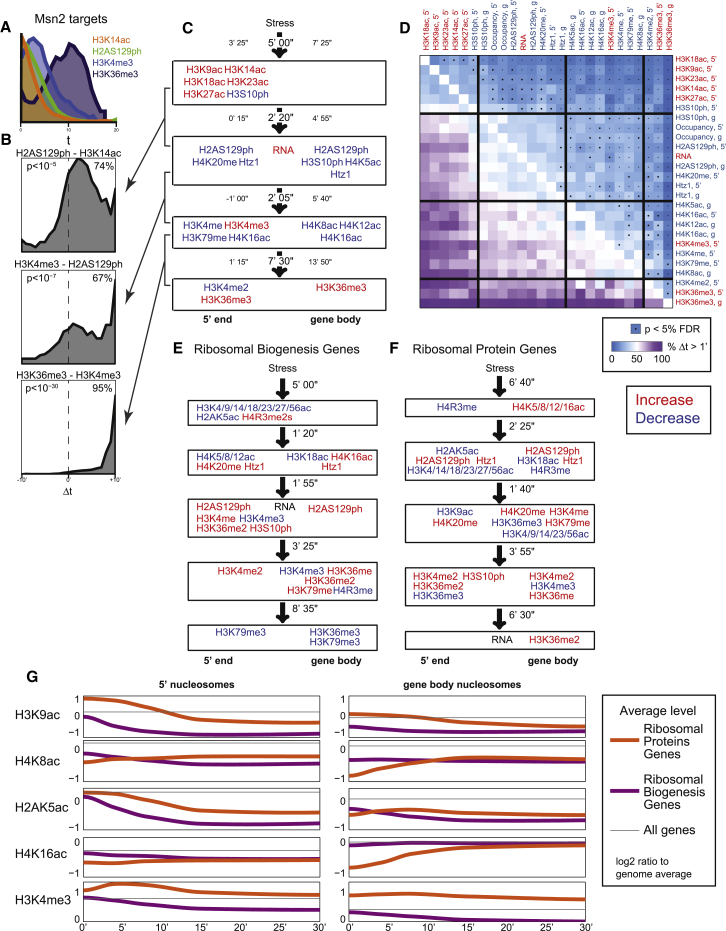
Covalent histone modifications are highly conserved and play multiple roles in eukaryotic transcription regulation. Here, we mapped 26 histone modifications genome-wide in exponentially growing yeast and during a dramatic transcriptional reprogramming-the response to diamide stress. We extend prior studies showing that steady-state histone modification patterns reflect genomic processes, especially transcription, and display limited combinatorial complexity. Interestingly, during the stress response we document a modest increase in the combinatorial complexity of histone modification space, resulting from roughly 3% of all nucleosomes transiently populating rare histone modification states. Most of these rare histone states result from differences in the kinetics of histone modification that transiently uncouple highly correlated marks, with slow histone methylation changes often lagging behind the more rapid acetylation changes. Explicit analysis of modification dynamics uncovers ordered sequences of events in gene activation and repression. Together, our results provide a comprehensive view of chromatin dynamics during a massive transcriptional upheaval.
Copyright © 2015 The Authors. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Adkins M.W., Carson J.J., English C.M., Ramey C.J., Tyler J.K. The histone chaperone anti-silencing function 1 stimulates the acetylation of newly synthesized histone H3 in S-phase. J. Biol. Chem. 2007;282:1334–1340. - PubMed
-
- Blecher-Gonen R., Barnett-Itzhaki Z., Jaitin D., Amann-Zalcenstein D., Lara-Astiaso D., Amit I. High-throughput chromatin immunoprecipitation for genome-wide mapping of in vivo protein-DNA interactions and epigenomic states. Nat. Protoc. 2013;8:539–554. - PubMed
-
- Celic I., Masumoto H., Griffith W.P., Meluh P., Cotter R.J., Boeke J.D., Verreault A. The sirtuins hst3 and Hst4p preserve genome integrity by controlling histone h3 lysine 56 deacetylation. Curr. Biol. 2006;16:1280–1289. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
