

Velaglucerase alfa (VPRIV) enzyme replacement therapy in patients with Gaucher disease: Long-term data from phase III clinical trials
- PMID: 25801797
- PMCID: PMC4654249
- DOI: 10.1002/ajh.24012
Velaglucerase alfa (VPRIV) enzyme replacement therapy in patients with Gaucher disease: Long-term data from phase III clinical trials
Abstract
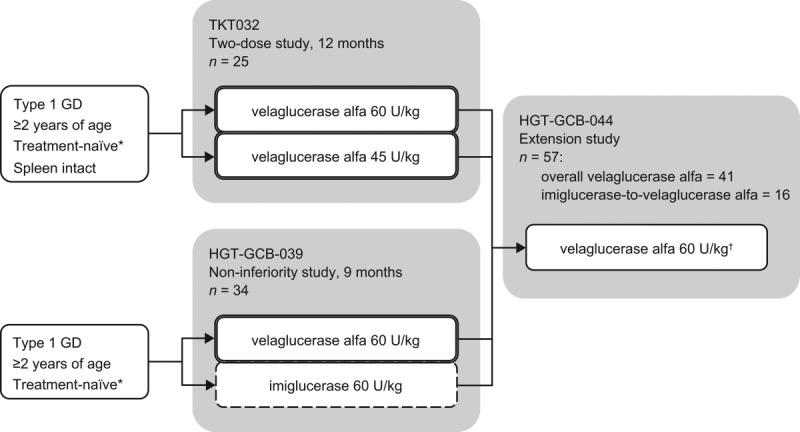
Type 1 Gaucher disease is an inherited lysosomal enzyme deficiency with variable age of symptom onset. Common presenting signs include thrombocytopenia, anemia, hepatosplenomegaly, bone abnormalities, and, additionally in children, growth failure. Fifty-seven patients aged 3-62 years at the baseline of two phase III trials for velaglucerase alfa treatment were enrolled in the single extension study. In the extension, they received every-other-week velaglucerase alfa intravenous infusions for 1.2-4.8 years at 60 U/kg, although 10 patients experienced dose reduction. No patient experienced a drug-related serious adverse event or withdrew due to an adverse event. One patient died following a convulsion that was reported as unrelated to the study drug. Only one patient tested positive for anti-velaglucerase alfa antibodies. Combining the experience of the initial phase III trials and the extension study, significant improvements were observed in the first 24 months from baseline in hematology variables, organ volumes, plasma biomarkers, and, in adults, the lumbar spine bone mineral density Z-score. Improvements were maintained over longer-term treatment. Velaglucerase alfa had a good long-term safety and tolerability profile, and patients continued to respond clinically, which is consistent with the results of the extension study to the phase I/II trial of velaglucerase alfa. EudraCT number 2008-001965-27; www.clinicaltrials.gov identifier NCT00635427.
© 2015 The Authors. American Journal of Hematology published by Wiley Periodicals, Inc.
Figures

Similar articles
-
Safety and efficacy results of switch from imiglucerase to velaglucerase alfa treatment in patients with type 1 Gaucher disease.Am J Hematol. 2015 Jul;90(7):592-7. doi: 10.1002/ajh.24007. Am J Hematol. 2015. PMID: 25776130 Clinical Trial.
-
Seven-year safety and efficacy with velaglucerase alfa for treatment-naïve adult patients with type 1 Gaucher disease.Am J Hematol. 2015 Jul;90(7):577-83. doi: 10.1002/ajh.24040. Epub 2015 May 28. Am J Hematol. 2015. PMID: 25903392 Free PMC article. Clinical Trial.
-
Velaglucerase alfa for the management of type 1 Gaucher disease.Clin Ther. 2012 Feb;34(2):259-71. doi: 10.1016/j.clinthera.2011.12.017. Epub 2012 Jan 20. Clin Ther. 2012. PMID: 22264444 Review.
-
Long-term velaglucerase alfa treatment in children with Gaucher disease type 1 naïve to enzyme replacement therapy or previously treated with imiglucerase.Mol Genet Metab. 2016 Feb;117(2):164-71. doi: 10.1016/j.ymgme.2015.05.012. Epub 2015 Jun 1. Mol Genet Metab. 2016. PMID: 26043810 Clinical Trial.
-
13,845 home therapy infusions with velaglucerase alfa exemplify safety of velaglucerase alfa and increased compliance to every-other-week intravenous enzyme replacement therapy for Gaucher disease.Blood Cells Mol Dis. 2015 Dec;55(4):415-8. doi: 10.1016/j.bcmd.2015.09.002. Epub 2015 Sep 21. Blood Cells Mol Dis. 2015. PMID: 26460268 Review.
Cited by
-
Twelve Years of the Gaucher Outcomes Survey (GOS): Insights, Achievements, and Lessons Learned from a Global Patient Registry.J Clin Med. 2024 Jun 19;13(12):3588. doi: 10.3390/jcm13123588. J Clin Med. 2024. PMID: 38930117 Free PMC article.
-
Global Incidence and Prevalence of Gaucher Disease: A Targeted Literature Review.J Clin Med. 2022 Dec 22;12(1):85. doi: 10.3390/jcm12010085. J Clin Med. 2022. PMID: 36614898 Free PMC article. Review.
-
Switching between Enzyme Replacement Therapies and Substrate Reduction Therapies in Patients with Gaucher Disease: Data from the Gaucher Outcome Survey (GOS).J Clin Med. 2022 Aug 31;11(17):5158. doi: 10.3390/jcm11175158. J Clin Med. 2022. PMID: 36079085 Free PMC article.
-
Long-term safety and effectiveness of velaglucerase alfa in Gaucher disease: 6-year interim analysis of a post-marketing surveillance in Japan.Orphanet J Rare Dis. 2021 Dec 4;16(1):502. doi: 10.1186/s13023-021-02119-2. Orphanet J Rare Dis. 2021. PMID: 34863216 Free PMC article.
-
Spotlight on taliglucerase alfa in the treatment of pediatric patients with type 1 Gaucher disease.Pediatric Health Med Ther. 2017 Jun 16;8:73-81. doi: 10.2147/PHMT.S93634. eCollection 2017. Pediatric Health Med Ther. 2017. PMID: 29388611 Free PMC article. Review.
References
-
- Mistry PK, Zimran A. Type 1 Gaucher disease—clinical features. In: Futerman AH, Zimran A, editors. Gaucher Disease. Boca Raton, FL: Taylor & Francis Group; 2007. p. 155–173.
-
- Wenstrup RJ, Kacena KA, Kaplan P, et al. Effect of enzyme replacement therapy with imiglucerase on BMD in type 1 Gaucher disease. J Bone Miner Res. 2007;22:119–126. - PubMed
-
- Andersson H, Kaplan P, Kacena K, et al. Eight-year clinical outcomes of long-term enzyme replacement therapy for 884 children with Gaucher disease type 1. Pediatrics. 2008;122:1182–1190. - PubMed
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical