

Any Way You Slice It-A Comparison of Confocal Microscopy Techniques
- PMID: 25802490
- PMCID: PMC4365987
- DOI: 10.7171/jbt.15-2602-003
Any Way You Slice It-A Comparison of Confocal Microscopy Techniques
Abstract
The confocal fluorescence microscope has become a popular tool for life sciences researchers, primarily because of its ability to remove blur from outside of the focal plane of the image. Several different kinds of confocal microscopes have been developed, each with advantages and disadvantages. This article will cover the grid confocal, classic confocal laser-scanning microscope (CLSM), the resonant scanning-CLSM, and the spinning-disk confocal microscope. The way each microscope technique works, the best applications the technique is suited for, the limitations of the technique, and new developments for each technology will be presented. Researchers who have access to a range of different confocal microscopes (e.g., through a local core facility) should find this paper helpful for choosing the best confocal technology for specific imaging applications. Others with funding to purchase an instrument should find the article helpful in deciding which technology is ideal for their area of research.
Keywords: 3D imaging; grid confocal; laser scanning; resonant scanning; spinning disk.
Figures

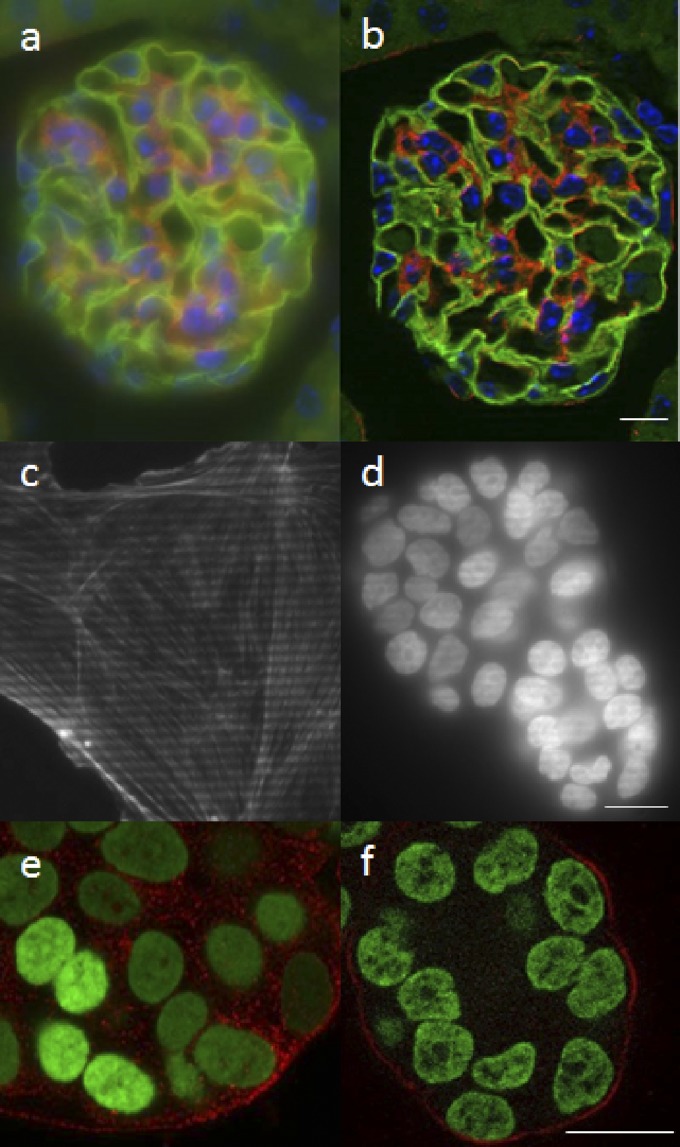
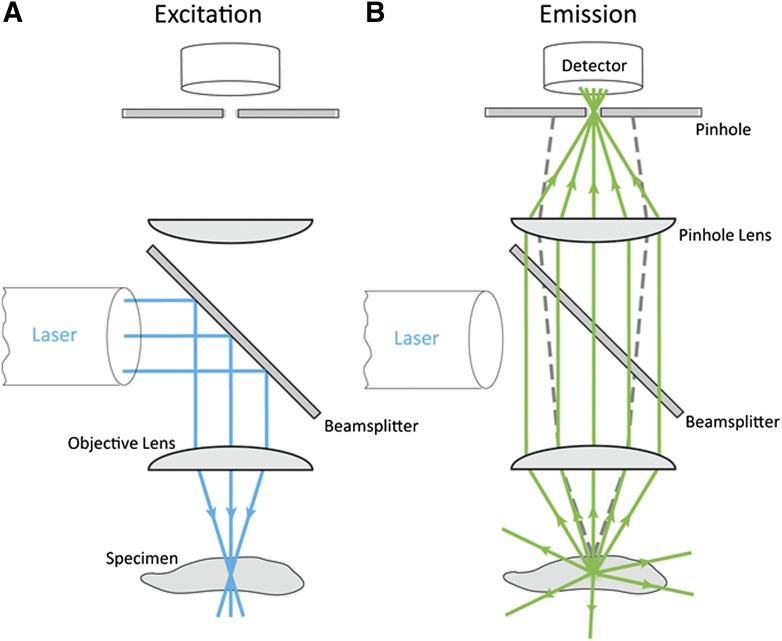
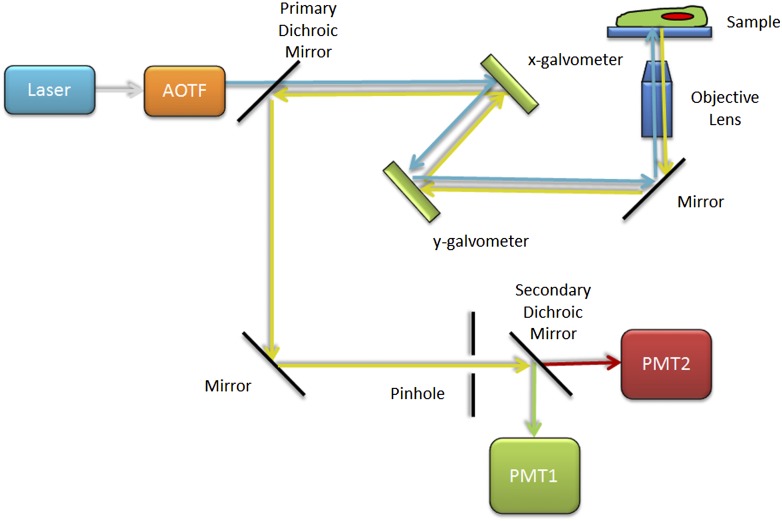

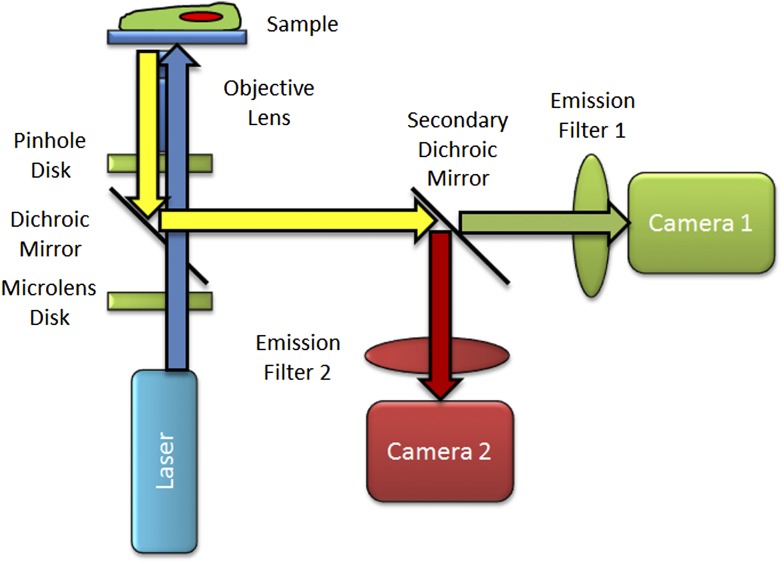

References
-
- Minsky M. Memoir on inventing the confocal scanning microscope. Scanning 1988;10:128–138.
-
- Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science 1990;248:73–76. - PubMed
-
- Galdeen SA, North AJ. Live cell fluorescence microscopy techniques. Methods Mol Biol 2011;769:205–222. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources