

Co-morbidity and systemic inflammation as drivers of cognitive decline: new experimental models adopting a broader paradigm in dementia research
- PMID: 25802557
- PMCID: PMC4369837
- DOI: 10.1186/s13195-015-0117-2
Co-morbidity and systemic inflammation as drivers of cognitive decline: new experimental models adopting a broader paradigm in dementia research
Abstract
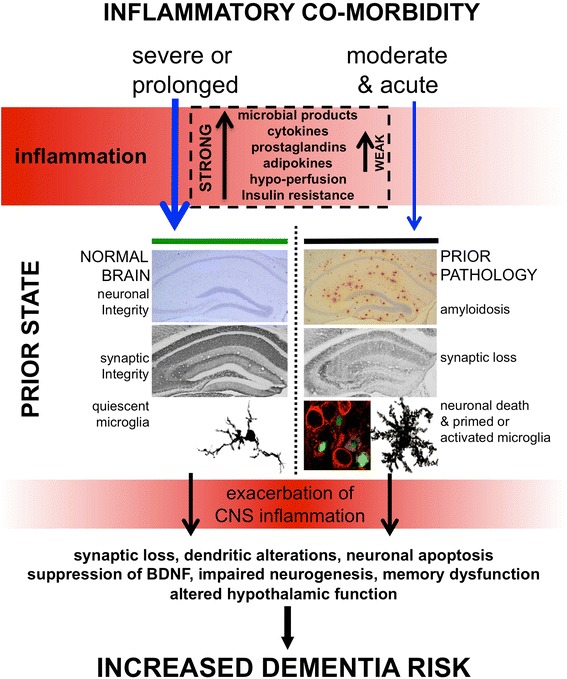
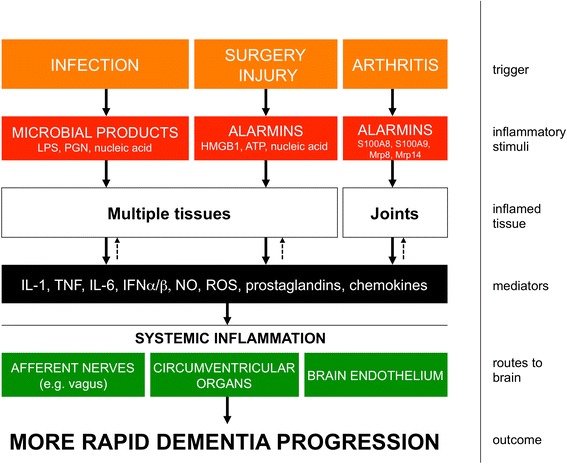
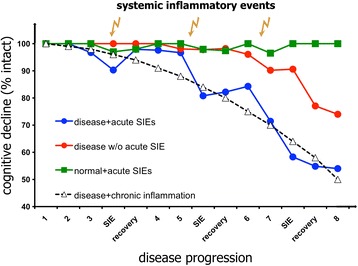
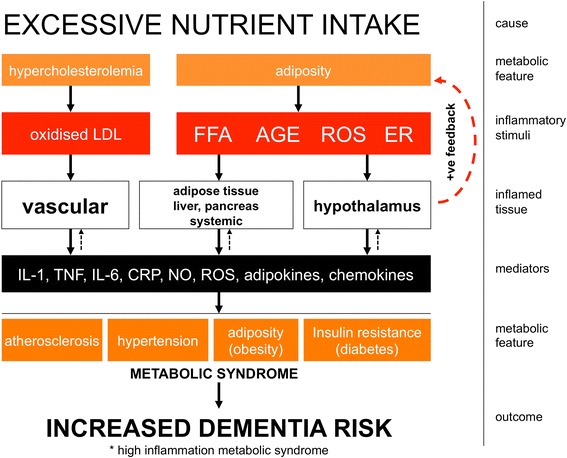
Dementia prevalence increases with age and Alzheimer's disease (AD) accounts for up to 75% of cases. However, significant variability and overlap exists in the extent of amyloid-β and Tau pathology in AD and non-demented populations and it is clear that other factors must influence progression of cognitive decline, perhaps independent of effects on amyloid pathology. Coupled with the failure of amyloid-clearing strategies to provide benefits for AD patients, it seems necessary to broaden the paradigm in dementia research beyond amyloid deposition and clearance. Evidence has emerged from alternative animal model approaches as well as clinical and population epidemiological studies that co-morbidities contribute significantly to neurodegeneration/cognitive decline and systemic inflammation has been a strong common theme in these approaches. We hypothesise, and discuss in this review, that a disproportionate inflammatory response to infection, injury or chronic peripheral disease is a key determinant of cognitive decline. We propose that detailed study of alternative models, which encompass acute and chronic systemic inflammatory co-morbidities, is an important priority for the field and we examine the cognitive consequences of several of these alternative experimental approaches. Experimental models of severe sepsis in normal animals or moderate acute systemic inflammation in animals with existing neurodegenerative pathology have uncovered roles for inflammatory mediators interleukin-1β, tumour necrosis factor-α, inducible nitric oxide synthase, complement, prostaglandins and NADPH oxidase in inflammation-induced cognitive dysfunction and neuronal death. Moreover, microglia are primed by existing neurodegenerative pathology to produce exaggerated responses to subsequent stimulation with bacterial lipopolysaccharide or other inflammatory stimuli and these insults drive acute dysfunction and negatively affect disease trajectory. Chronic co-morbidities, such as arthritis, atherosclerosis, obesity and diabetes, are risk factors for subsequent dementia and those with high inflammatory status are particularly at risk. Models of chronic co-morbidities, and indeed low grade systemic inflammation in the absence of specific pathology, indicate that interleukin-1β, tumour necrosis factor-α and other inflammatory mediators drive insulin resistance, hypothalamic dysfunction, impaired neurogenesis and cognitive function and impact on functional decline. Detailed study of these pathways will uncover important mechanisms of peripheral inflammation-driven cognitive decline and are already driving clinical initiatives to mitigate AD progression through minimising systemic inflammation.
Figures
References
-
- Neuropathology Group Medical Research Council Cognitive Function and Aging Study. Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Lancet. 2001;357:169–75. - PubMed
-
- Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, et al. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–23. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
