

Variability in pathogenicity prediction programs: impact on clinical diagnostics
- PMID: 25802880
- PMCID: PMC4367082
- DOI: 10.1002/mgg3.116
Variability in pathogenicity prediction programs: impact on clinical diagnostics
Abstract
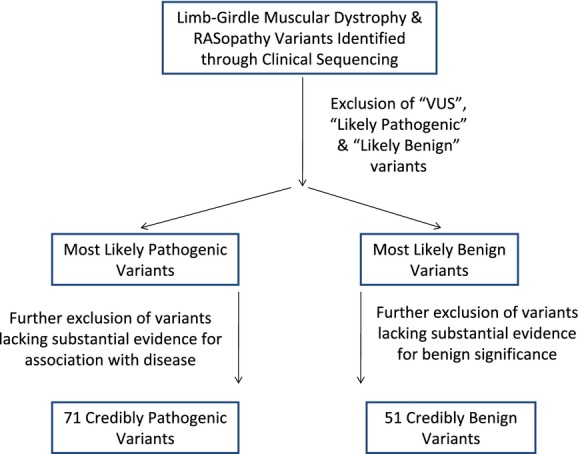
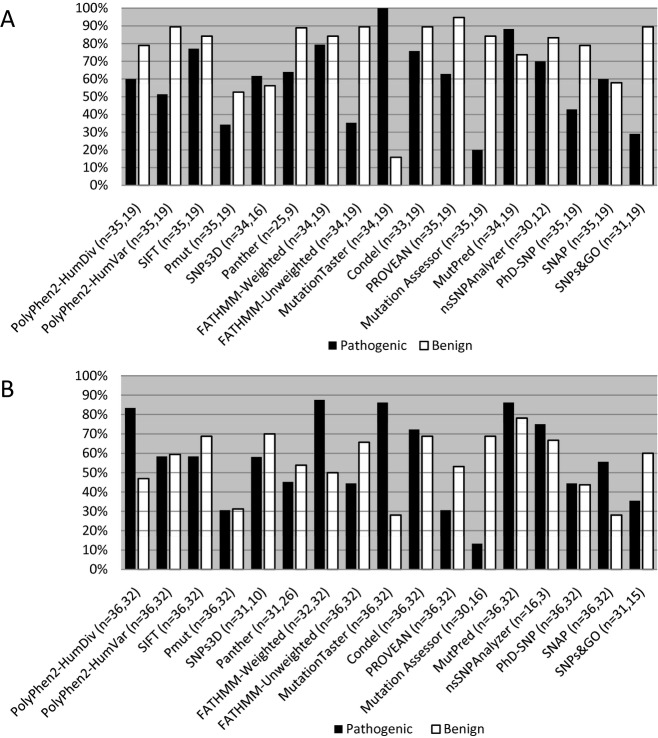
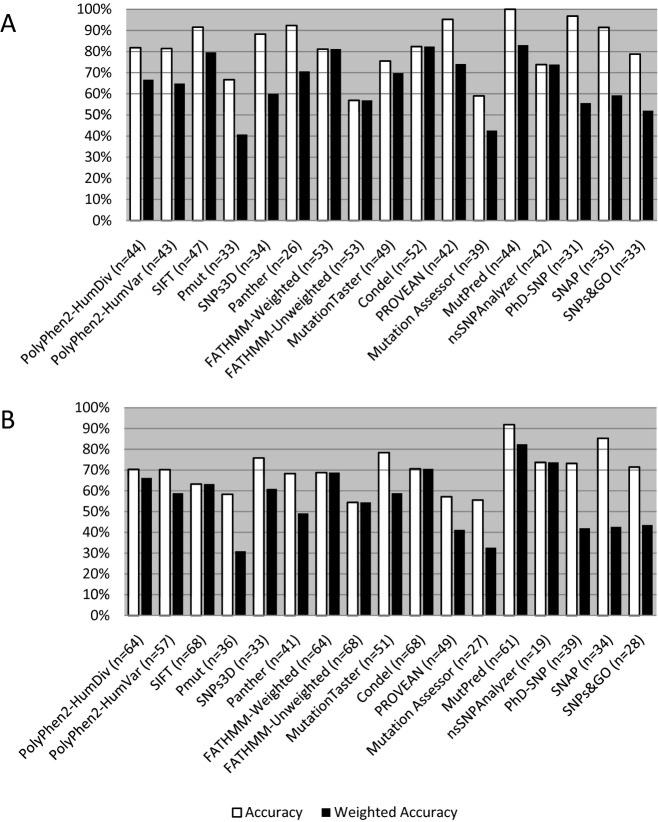
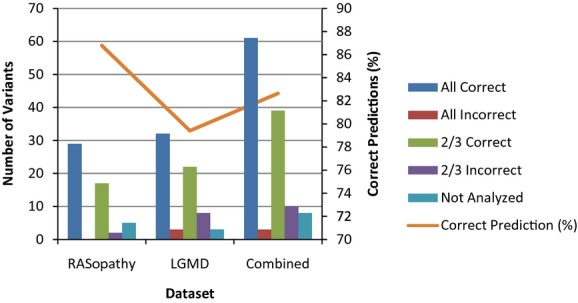
Current practice by clinical diagnostic laboratories is to utilize online prediction programs to help determine the significance of novel variants in a given gene sequence. However, these programs vary widely in their methods and ability to correctly predict the pathogenicity of a given sequence change. The performance of 17 publicly available pathogenicity prediction programs was assayed using a dataset consisting of 122 credibly pathogenic and benign variants in genes associated with the RASopathy family of disorders and limb-girdle muscular dystrophy. Performance metrics were compared between the programs to determine the most accurate program for loss-of-function and gain-of-function mechanisms. No one program correctly predicted the pathogenicity of all variants analyzed. A major hindrance to the analysis was the lack of output from a significant portion of the programs. The best performer was MutPred, which had a weighted accuracy of 82.6% in the full dataset. Surprisingly, combining the results of the top three programs did not increase the ability to predict pathogenicity over the top performer alone. As the increasing number of sequence changes in larger datasets will require interpretation, the current study demonstrates that extreme caution must be taken when reporting pathogenicity based on statistical online protein prediction programs in the absence of functional studies.
Keywords: Diagnostics; pathogenicity; prediction; sequencing; variants.
Figures
References
-
- Calabrese R, Capriotti E, Fariselli P, Martelli PL. Casadio R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum. Mutat. 2009;30:1237–1244. - PubMed
-
- Capriotti E, Calabrese R. Casadio R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics. 2006;22:2729–2734. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
