

Conservatism and novelty in the genetic architecture of adaptation in Heliconius butterflies
- PMID: 25806542
- PMCID: PMC4815517
- DOI: 10.1038/hdy.2015.22
Conservatism and novelty in the genetic architecture of adaptation in Heliconius butterflies
Abstract
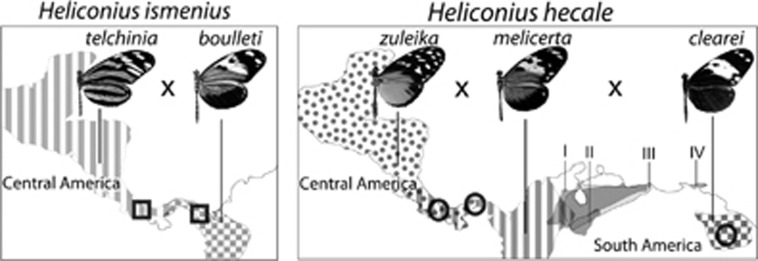
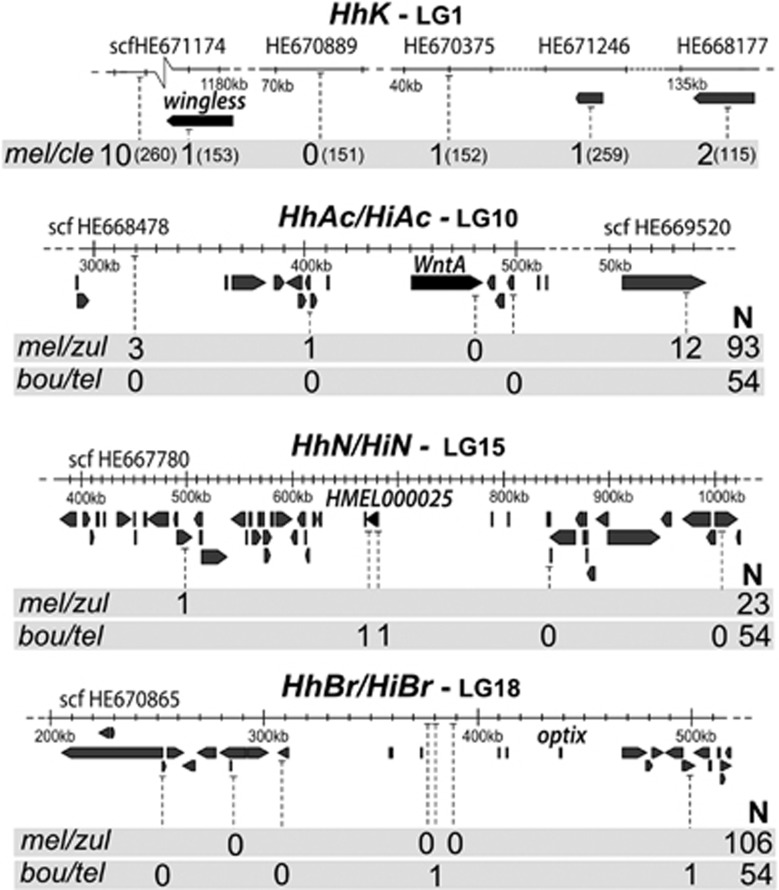
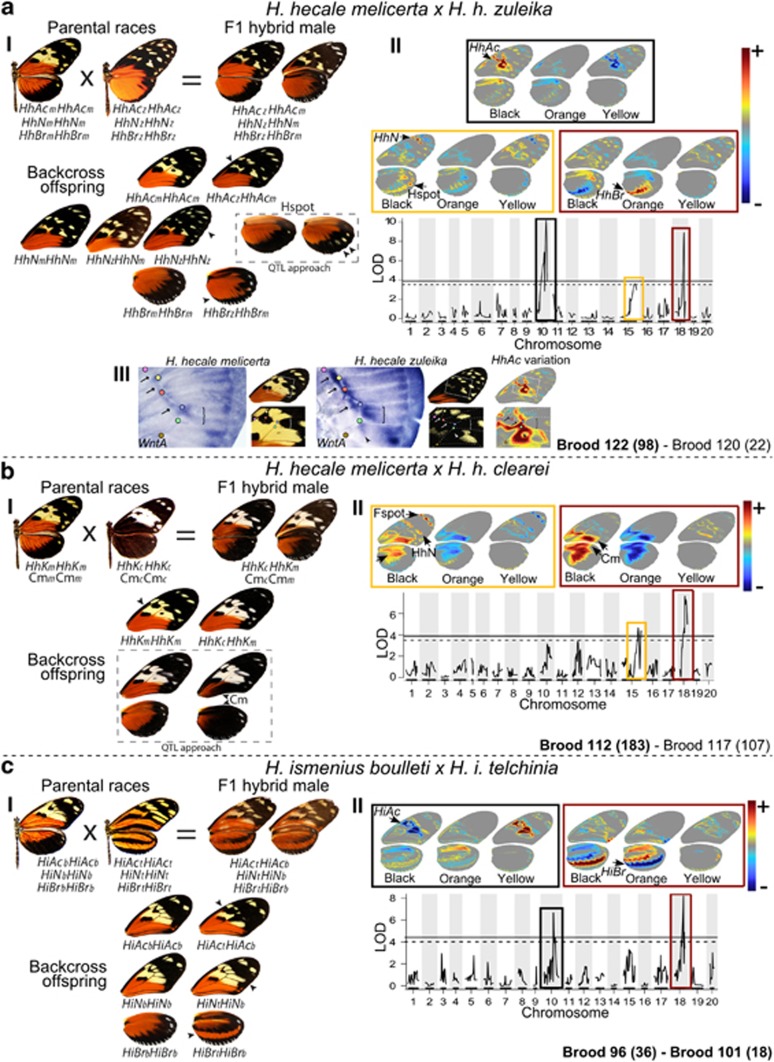
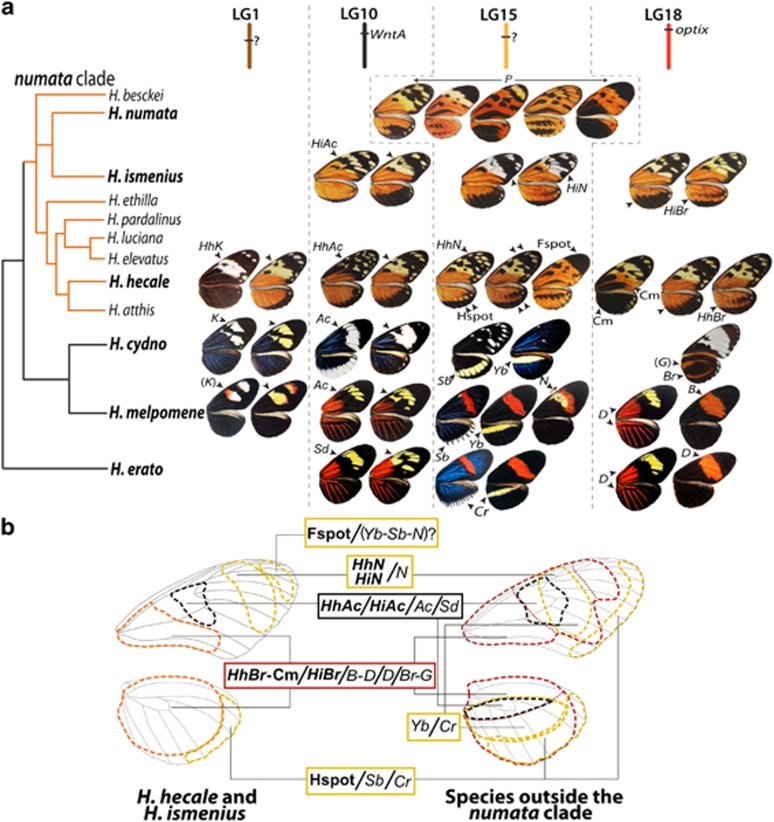
Understanding the genetic architecture of adaptive traits has been at the centre of modern evolutionary biology since Fisher; however, evaluating how the genetic architecture of ecologically important traits influences their diversification has been hampered by the scarcity of empirical data. Now, high-throughput genomics facilitates the detailed exploration of variation in the genome-to-phenotype map among closely related taxa. Here, we investigate the evolution of wing pattern diversity in Heliconius, a clade of neotropical butterflies that have undergone an adaptive radiation for wing-pattern mimicry and are influenced by distinct selection regimes. Using crosses between natural wing-pattern variants, we used genome-wide restriction site-associated DNA (RAD) genotyping, traditional linkage mapping and multivariate image analysis to study the evolution of the architecture of adaptive variation in two closely related species: Heliconius hecale and H. ismenius. We implemented a new morphometric procedure for the analysis of whole-wing pattern variation, which allows visualising spatial heatmaps of genotype-to-phenotype association for each quantitative trait locus separately. We used the H. melpomene reference genome to fine-map variation for each major wing-patterning region uncovered, evaluated the role of candidate genes and compared genetic architectures across the genus. Our results show that, although the loci responding to mimicry selection are highly conserved between species, their effect size and phenotypic action vary throughout the clade. Multilocus architecture is ancestral and maintained across species under directional selection, whereas the single-locus (supergene) inheritance controlling polymorphism in H. numata appears to have evolved only once. Nevertheless, the conservatism in the wing-patterning toolkit found throughout the genus does not appear to constrain phenotypic evolution towards local adaptive optima.
Figures
References
-
- Arendt J, Reznick D. (2008). Convergence and parallelism reconsidered: what have we learned about the genetics of adaptation? Trends Ecol Evol 23: 26–32. - PubMed
-
- Barton NH. (1995). A general model for the evolution of recombination. Genet Res 65: 123–145. - PubMed
-
- Baxter SW, Johnston SE, Jiggins CD. (2008). Butterfly speciation and the distribution of gene effect sizes fixed during adaptation. Heredity 102: 57–65. - PubMed
-
- Broman K, Sen S. (2009) A Guide to QTL Mapping with R/qtl. Springer: New York.
-
- Broman KW, Wu H, Sen S, Churchill GA. (2003). R/qtl: QTL mapping in experimental crosses. Bioinforma Oxf Engl 19: 889–890. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
