

A new asynchronous parallel algorithm for inferring large-scale gene regulatory networks
- PMID: 25807392
- PMCID: PMC4373852
- DOI: 10.1371/journal.pone.0119294
A new asynchronous parallel algorithm for inferring large-scale gene regulatory networks
Abstract
The reconstruction of gene regulatory networks (GRNs) from high-throughput experimental data has been considered one of the most important issues in systems biology research. With the development of high-throughput technology and the complexity of biological problems, we need to reconstruct GRNs that contain thousands of genes. However, when many existing algorithms are used to handle these large-scale problems, they will encounter two important issues: low accuracy and high computational cost. To overcome these difficulties, the main goal of this study is to design an effective parallel algorithm to infer large-scale GRNs based on high-performance parallel computing environments. In this study, we proposed a novel asynchronous parallel framework to improve the accuracy and lower the time complexity of large-scale GRN inference by combining splitting technology and ordinary differential equation (ODE)-based optimization. The presented algorithm uses the sparsity and modularity of GRNs to split whole large-scale GRNs into many small-scale modular subnetworks. Through the ODE-based optimization of all subnetworks in parallel and their asynchronous communications, we can easily obtain the parameters of the whole network. To test the performance of the proposed approach, we used well-known benchmark datasets from Dialogue for Reverse Engineering Assessments and Methods challenge (DREAM), experimentally determined GRN of Escherichia coli and one published dataset that contains more than 10 thousand genes to compare the proposed approach with several popular algorithms on the same high-performance computing environments in terms of both accuracy and time complexity. The numerical results demonstrate that our parallel algorithm exhibits obvious superiority in inferring large-scale GRNs.
Conflict of interest statement
Figures
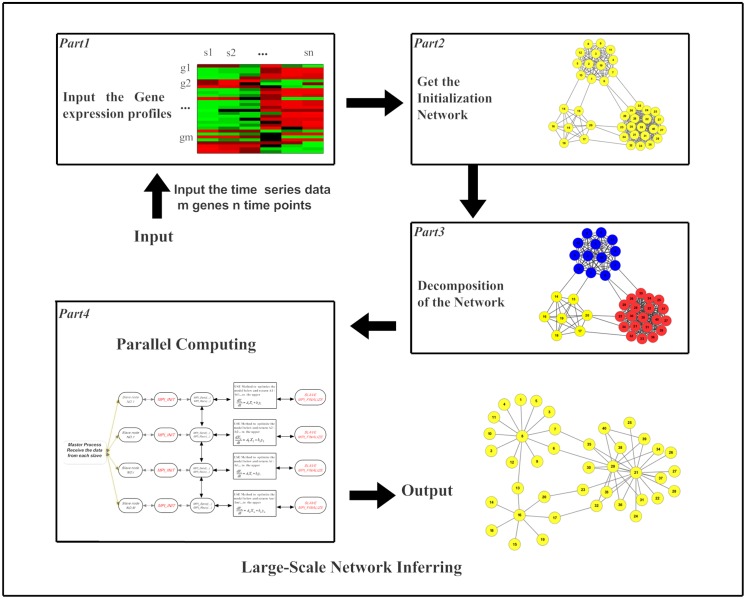
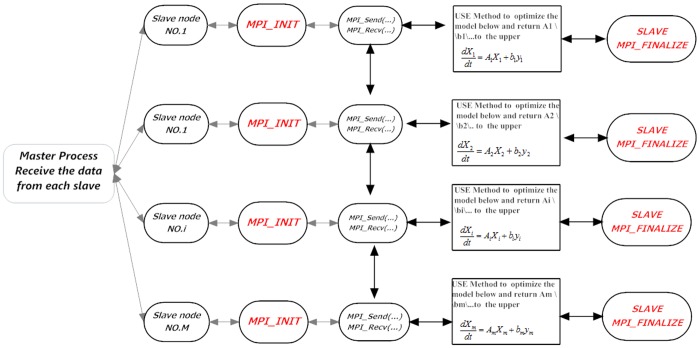
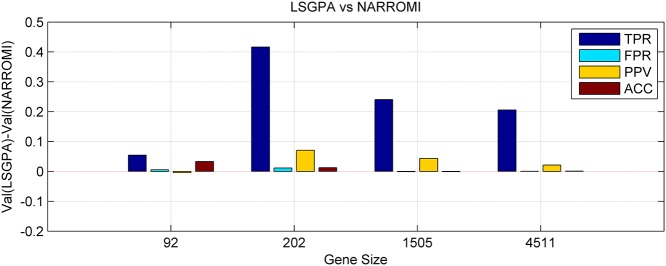
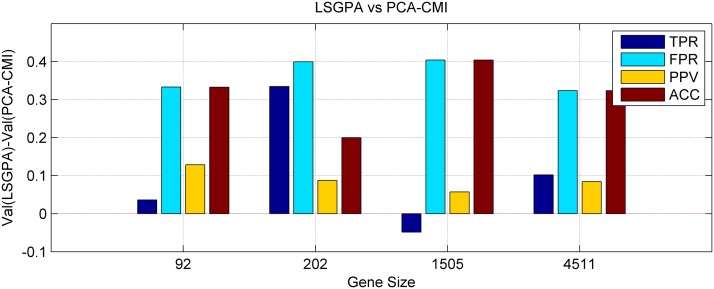
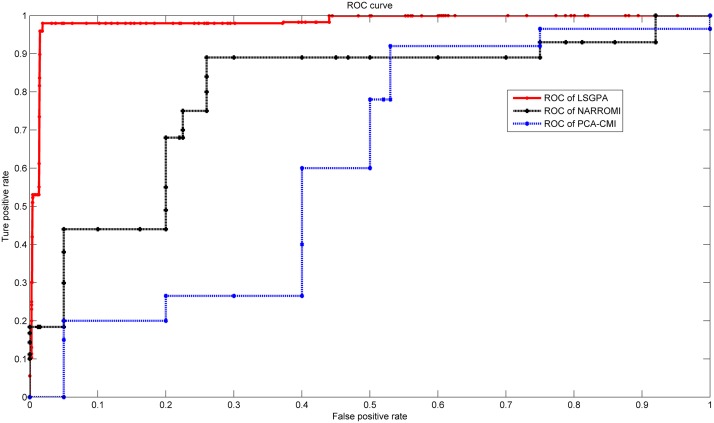
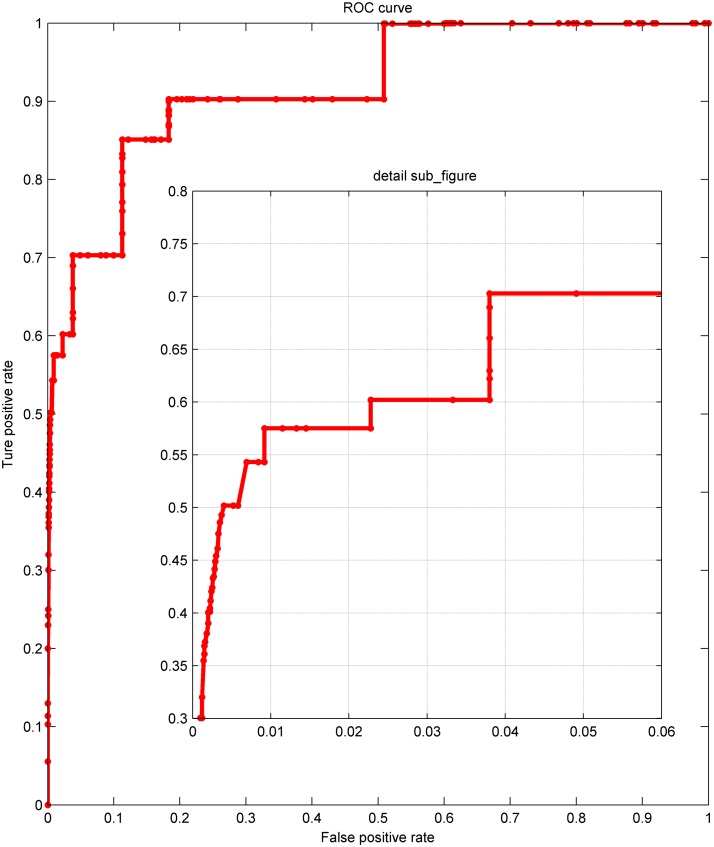
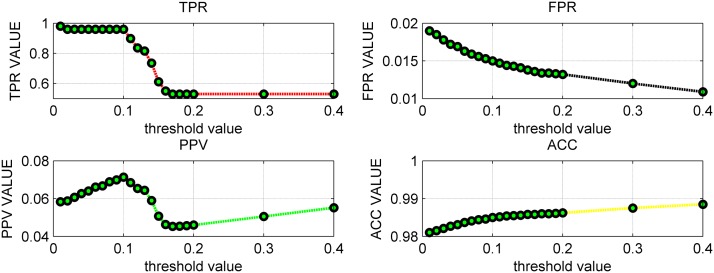
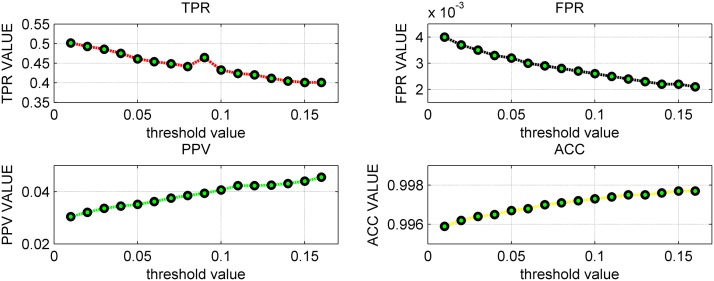
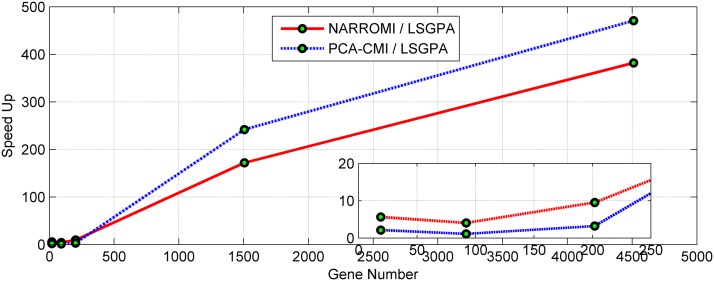
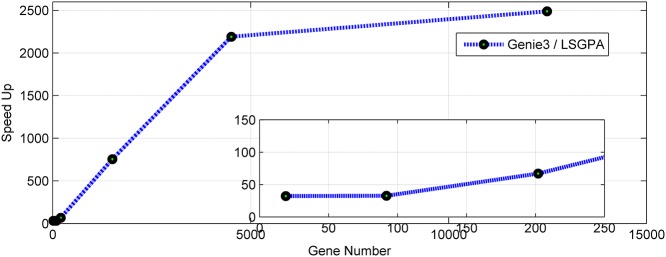
References
-
- Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, Califano A, et al. Reverse engineering of regulatory networks in human B cells. Nat Genet. 2005; 37: 382–390. - PubMed
-
- Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005; 4: Article17 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
