

Function and Mechanisms of Autophagy in Brain and Spinal Cord Trauma
- PMID: 25808205
- PMCID: PMC4545370
- DOI: 10.1089/ars.2015.6306
Function and Mechanisms of Autophagy in Brain and Spinal Cord Trauma
Abstract
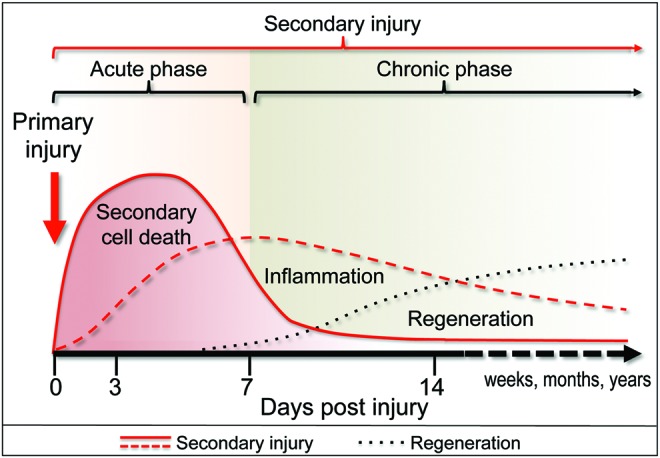
Significance: Traumatic brain injury (TBI) and spinal cord injury (SCI) are major causes of death and long-term disability worldwide. Despite important pathophysiological differences between these disorders, in many respects, mechanisms of injury are similar. During both TBI and SCI, some cells are directly mechanically injured, but more die as a result of injury-induced biochemical changes (secondary injury). Autophagy, a lysosome-dependent cellular degradation pathway with neuroprotective properties, has been implicated both clinically and experimentally in the delayed response to TBI and SCI. However, until recently, its mechanisms and function remained unknown, reflecting in part the difficulty of isolating autophagic processes from ongoing cell death and other cellular events.
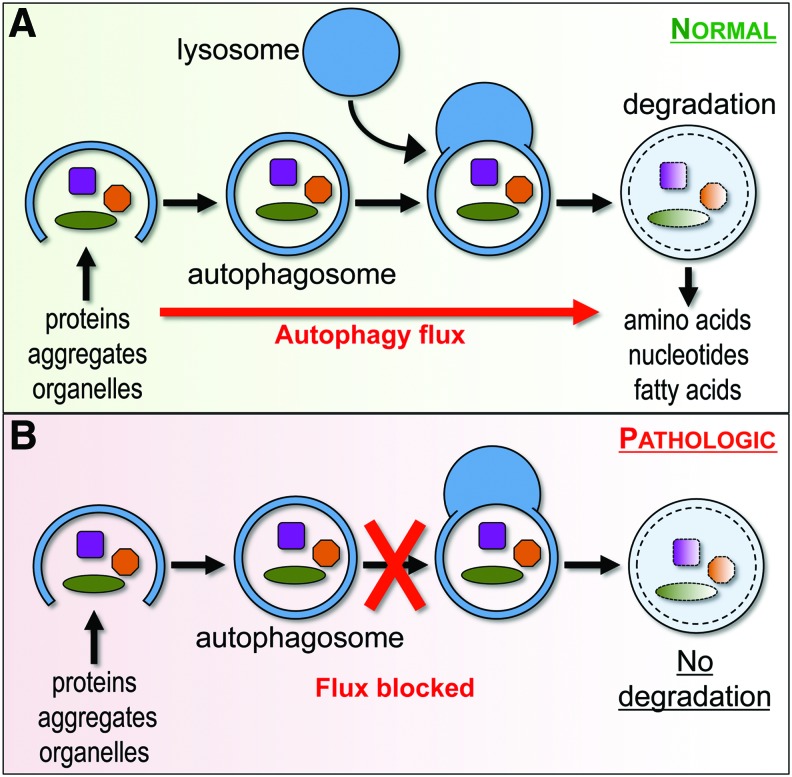
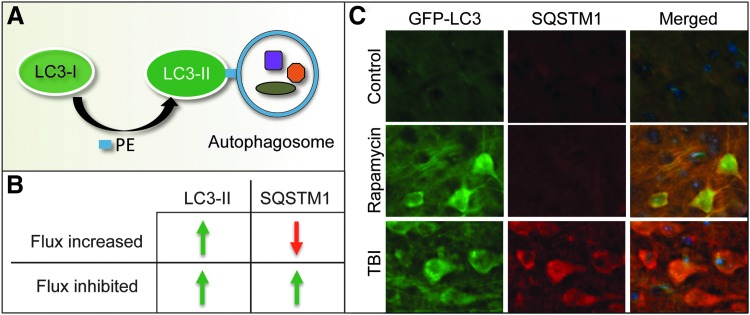
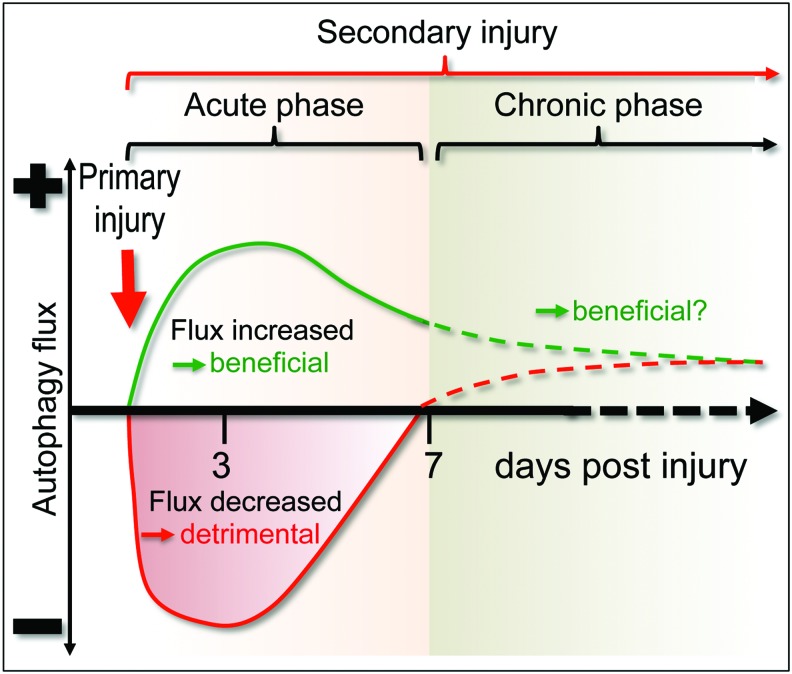
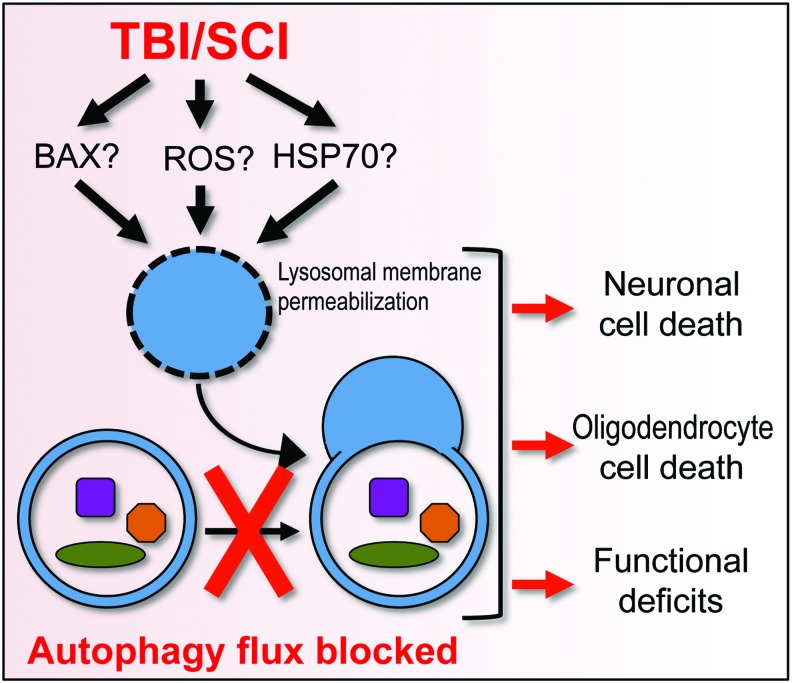
Recent advances: Emerging data suggest that depending on the location and severity of traumatic injury, autophagy flux--defined as the progress of cargo through the autophagy system and leading to its degradation--may be either increased or decreased after central nervous system trauma.
Critical issues: While increased autophagy flux may be protective after mild injury, after more severe trauma inhibition of autophagy flux may contribute to neuronal cell death, indicating disruption of autophagy as a part of the secondary injury mechanism.
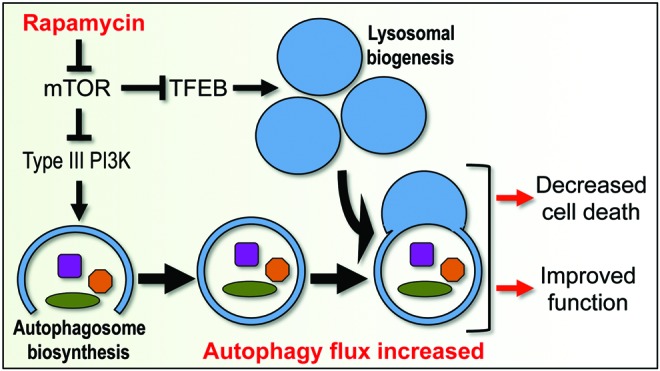
Future directions: Augmentation and/or restoration of autophagy flux may provide a potential therapeutic target for treatment of TBI and SCI. Development of those treatments will require thorough characterization of changes in autophagy flux, its mechanisms and function over time after injury.
Figures
References
-
- Injury Prevention and Control: Traumatic Brain Injury Center for Disease Control and Prevention. http://cdc.gov/traumaticbraininjury/2014 (Accessed date November/26/2014)
-
- Spinal Cord Injury (SCI): Fact Sheet Center for Disease Control and Prevention. http://cdc.gov/TraumaticBrainInjury/scifacts.html 2014. (Accessed date November/26/2014)
-
- Beattie MS, Hermann GE, Rogers RC, and Bresnahan JC. Cell death in models of spinal cord injury. Prog Brain Res 137: 37–47, 2002 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
