

An integrated map of HIV genome-wide variation from a population perspective
- PMID: 25808207
- PMCID: PMC4358901
- DOI: 10.1186/s12977-015-0148-6
An integrated map of HIV genome-wide variation from a population perspective
Abstract
Background: The HIV pandemic is characterized by extensive genetic variability, which has challenged the development of HIV drugs and vaccines. Although HIV genomes have been classified into different types, groups, subtypes and recombinants, a comprehensive study that maps HIV genome-wide diversity at the population level is still lacking to date. This study aims to characterize HIV genomic diversity in large-scale sequence populations, and to identify driving factors that shape HIV genome diversity.
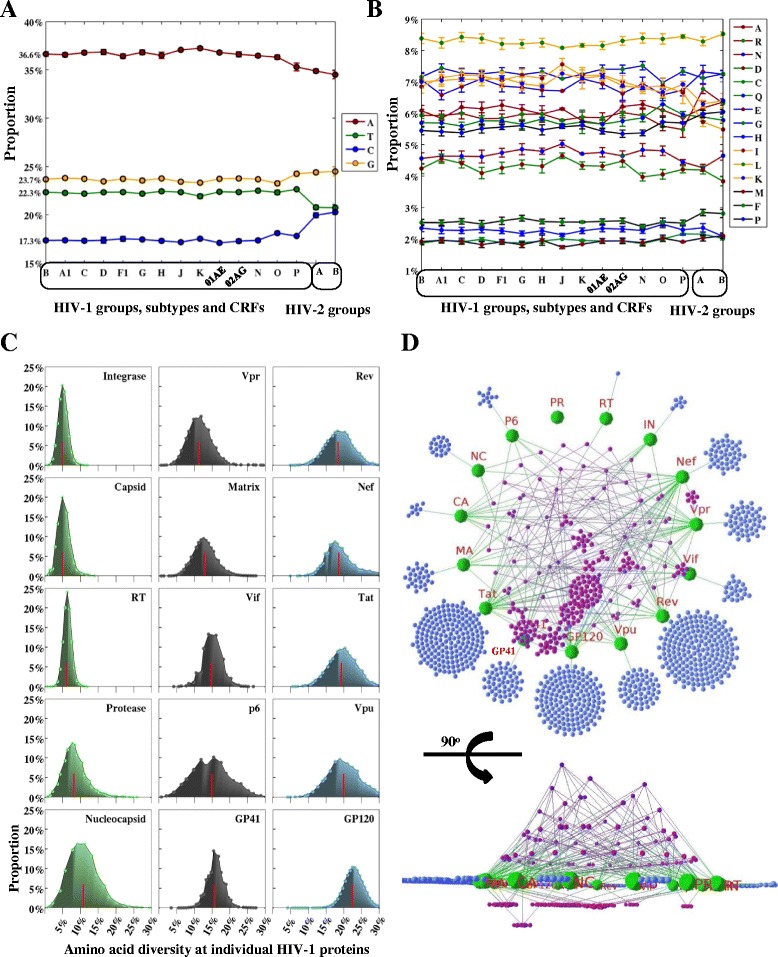
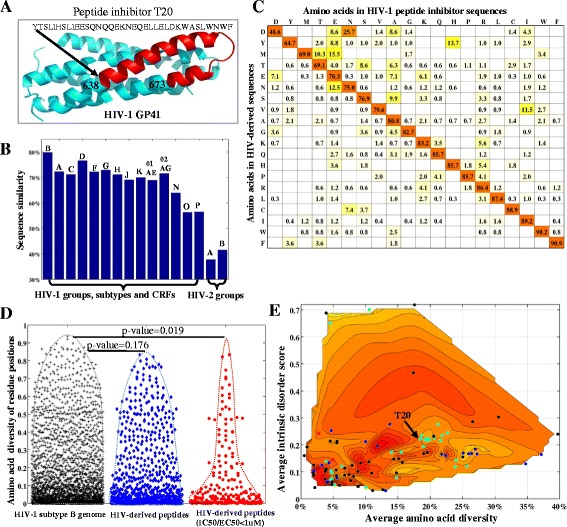
Results: A total of 2996 full-length genomic sequences from 1705 patients infected with 16 major HIV groups, subtypes and circulating recombinant forms (CRFs) were analyzed along with structural, immunological and peptide inhibitor information. Average nucleotide diversity of HIV genomes was almost 50% between HIV-1 and HIV-2 types, 37.5% between HIV-1 groups, 14.7% between HIV-1 subtypes, 8.2% within individual HIV-1 subtypes and less than 1% within single patients. Along the HIV genome, diversity patterns and compositions of nucleotides and amino acids were highly similar across different groups, subtypes and CRFs. Current HIV-derived peptide inhibitors were predominantly derived from conserved, solvent accessible and intrinsically ordered structures in the HIV-1 subtype B genome. We identified these conserved regions in Capsid, Nucleocapsid, Protease, Integrase, Reverse transcriptase, Vpr and the GP41 N terminus as potential drug targets. In the analysis of factors that impact HIV-1 genomic diversity, we focused on protein multimerization, immunological constraints and HIV-human protein interactions. We found that amino acid diversity in monomeric proteins was higher than in multimeric proteins, and diversified positions were preferably located within human CD4 T cell and antibody epitopes. Moreover, intrinsic disorder regions in HIV-1 proteins coincided with high levels of amino acid diversity, facilitating a large number of interactions between HIV-1 and human proteins.
Conclusions: This first large-scale analysis provided a detailed mapping of HIV genomic diversity and highlighted drug-target regions conserved across different groups, subtypes and CRFs. Our findings suggest that, in addition to the impact of protein multimerization and immune selective pressure on HIV-1 diversity, HIV-human protein interactions are facilitated by high variability within intrinsically disordered structures.
Figures




References
Publication types
MeSH terms
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
