

Development of Resistance to EGFR-Targeted Therapy in Malignant Glioma Can Occur through EGFR-Dependent and -Independent Mechanisms
- PMID: 25808866
- PMCID: PMC4433602
- DOI: 10.1158/0008-5472.CAN-14-3122
Development of Resistance to EGFR-Targeted Therapy in Malignant Glioma Can Occur through EGFR-Dependent and -Independent Mechanisms
Abstract
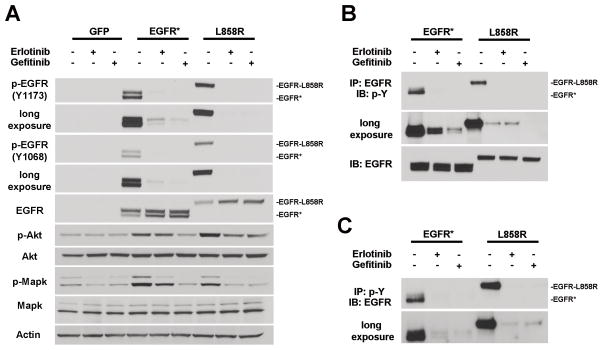
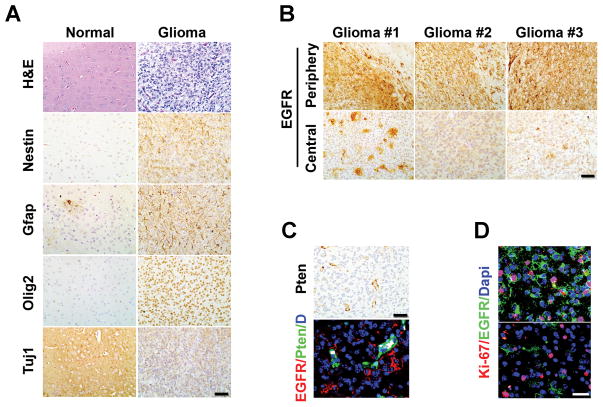
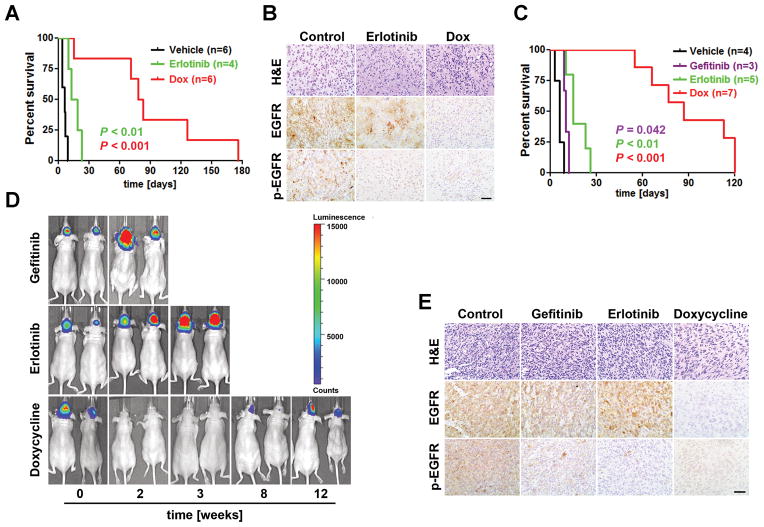
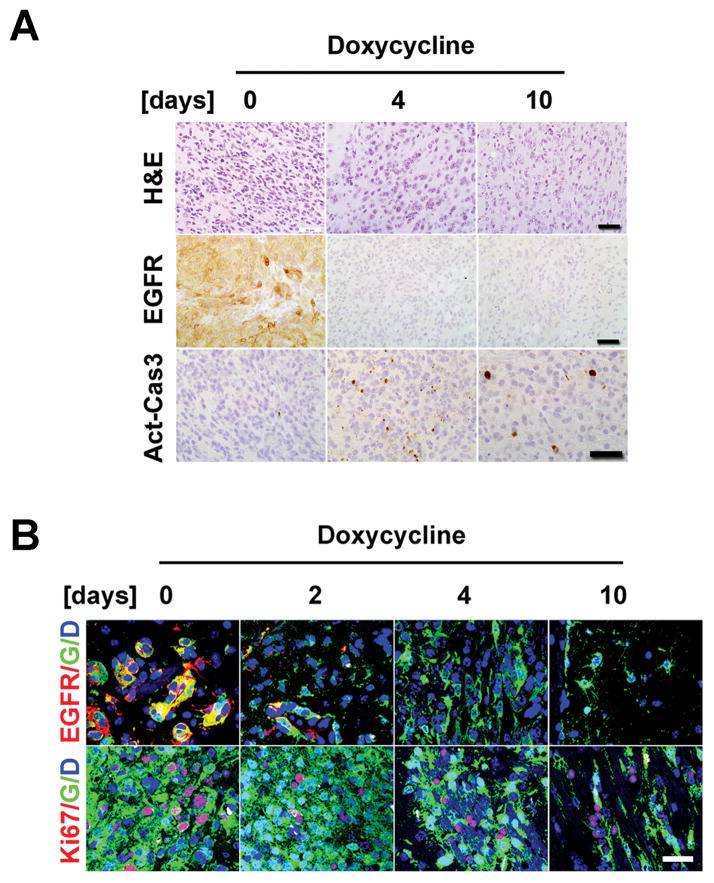
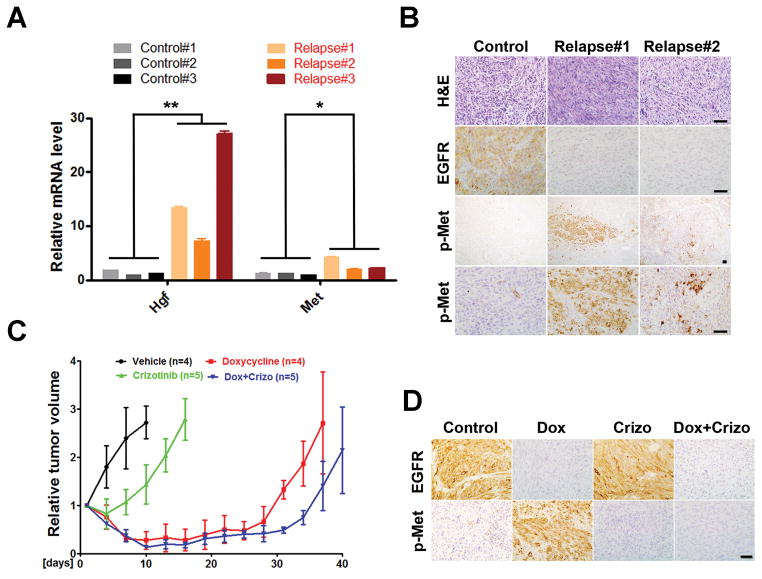
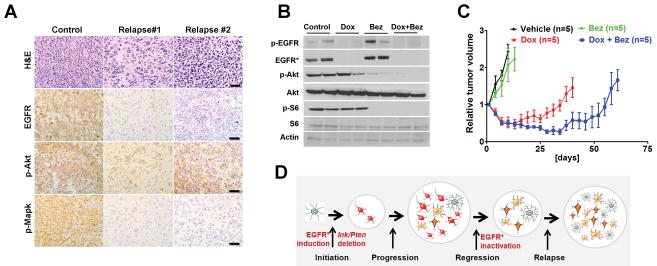
Epidermal growth factor receptor (EGFR) is highly amplified, mutated, and overexpressed in human malignant gliomas. Despite its prevalence and growth-promoting functions, therapeutic strategies to inhibit EGFR kinase activity have not been translated into profound beneficial effects in glioma clinical trials. To determine the roles of oncogenic EGFR signaling in gliomagenesis and tumor maintenance, we generated a novel glioma mouse model driven by inducible expression of a mutant EGFR (EGFR*). Using combined genetic and pharmacologic interventions, we revealed that EGFR*-driven gliomas were insensitive to EGFR tyrosine kinase inhibitors, although they could efficiently inhibit EGFR* autophosphorylation in vitro and in vivo. This is in contrast with the genetic suppression of EGFR* induction that led to significant tumor regression and prolonged animal survival. However, despite their initial response to genetic EGFR* extinction, all tumors would relapse and propagate independent of EGFR*. We further showed that EGFR*-independent tumor cells existed prior to treatment and were responsible for relapse following genetic EGFR* suppression. And, the addition of a PI3K/mTOR inhibitor could significantly delay relapse and prolong animal survival. Our findings shed mechanistic insight into EGFR drug resistance in glioma and provide a platform to test therapies targeting aberrant EGFR signaling in this setting.
©2015 American Association for Cancer Research.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
