

Failed Tubule Recovery, AKI-CKD Transition, and Kidney Disease Progression
- PMID: 25810494
- PMCID: PMC4520181
- DOI: 10.1681/ASN.2015010006
Failed Tubule Recovery, AKI-CKD Transition, and Kidney Disease Progression
Abstract
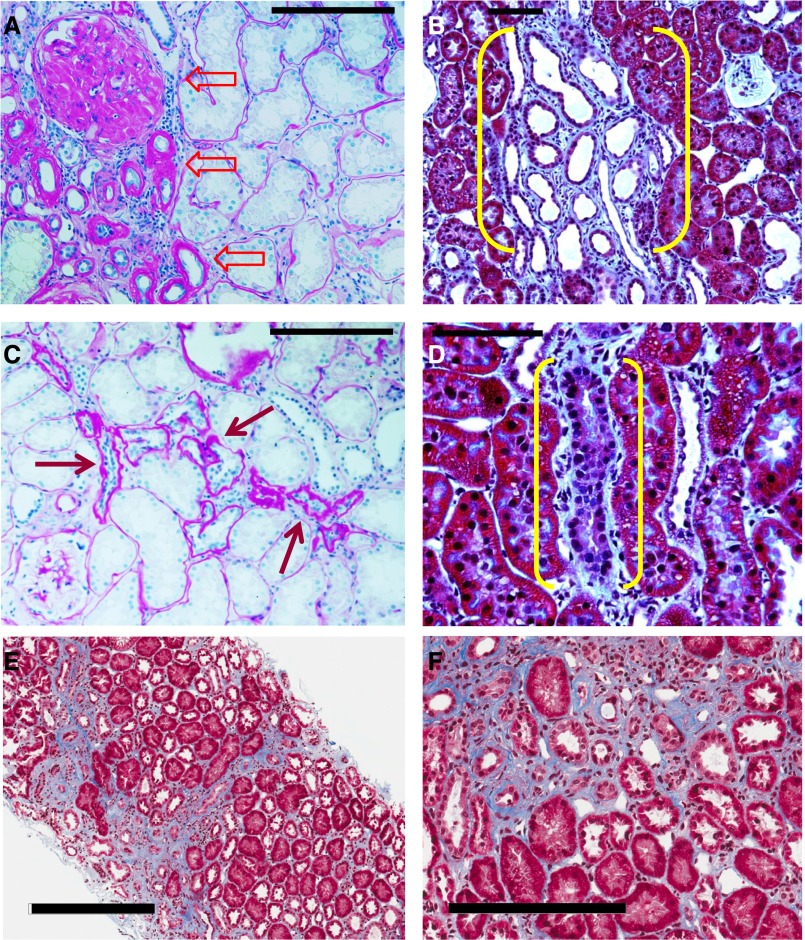
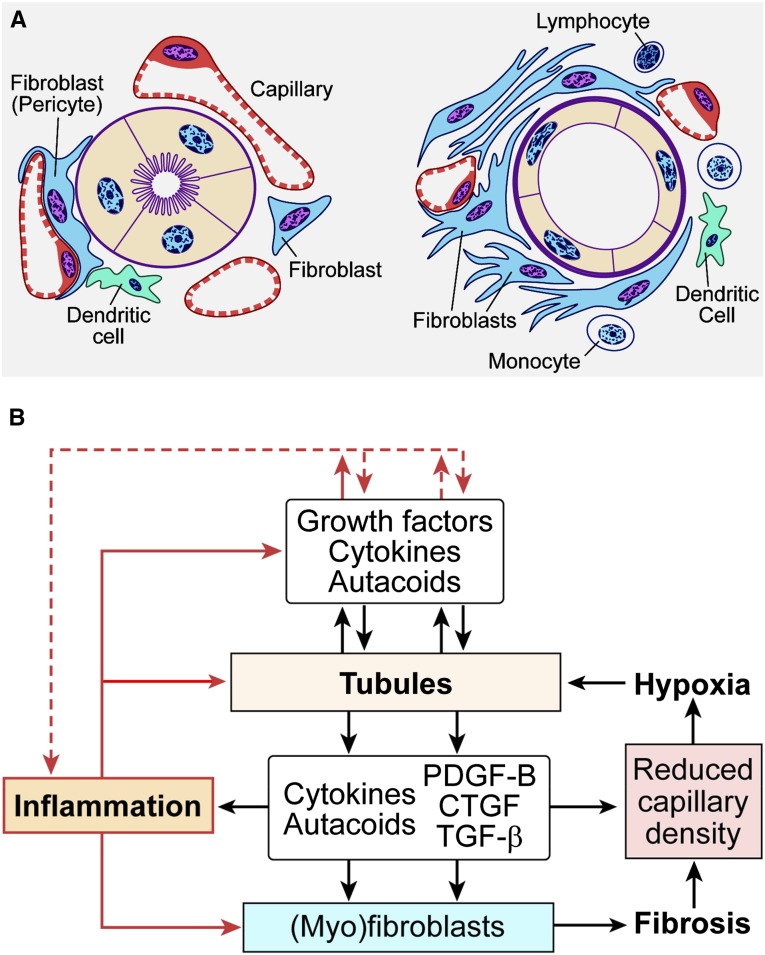
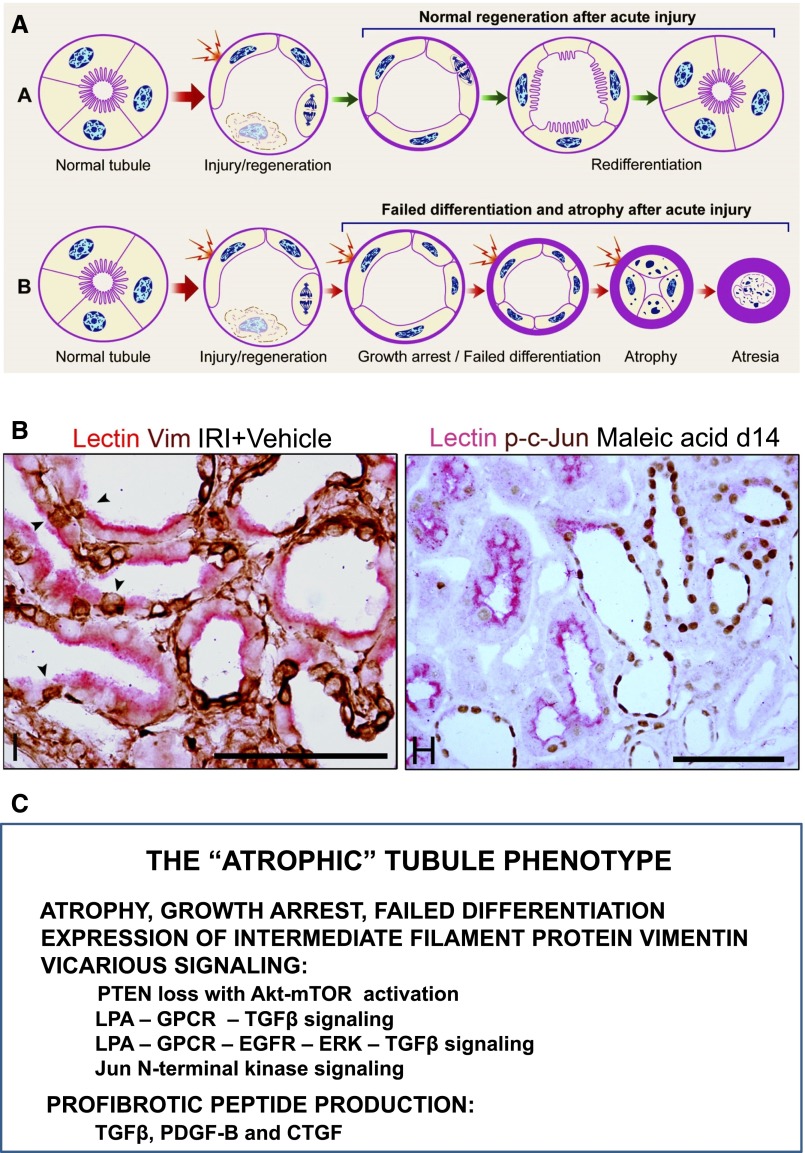
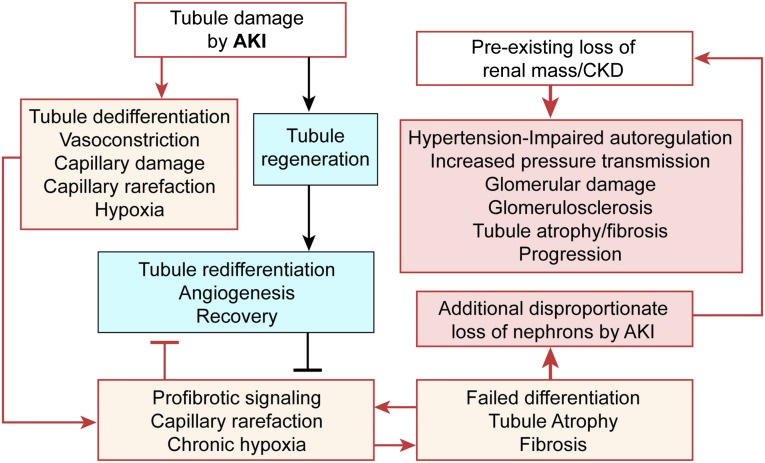
The transition of AKI to CKD has major clinical significance. As reviewed here, recent studies show that a subpopulation of dedifferentiated, proliferating tubules recovering from AKI undergo pathologic growth arrest, fail to redifferentiate, and become atrophic. These abnormal tubules exhibit persistent, unregulated, and progressively increasing profibrotic signaling along multiple pathways. Paracrine products derived therefrom perturb normal interactions between peritubular capillary endothelium and pericyte-like fibroblasts, leading to myofibroblast transformation, proliferation, and fibrosis as well as capillary disintegration and rarefaction. Although signals from injured endothelium and inflammatory/immune cells also contribute, tubule injury alone is sufficient to produce the interstitial pathology required for fibrosis. Localized hypoxia produced by microvascular pathology may also prevent tubule recovery. However, fibrosis is not intrinsically progressive, and microvascular pathology develops strictly around damaged tubules; thus, additional deterioration of kidney structure after the transition of AKI to CKD requires new acute injury or other mechanisms of progression. Indeed, experiments using an acute-on-chronic injury model suggest that additional loss of parenchyma caused by failed repair of AKI in kidneys with prior renal mass reduction triggers hemodynamically mediated processes that damage glomeruli to cause progression. Continued investigation of these pathologic mechanisms should reveal options for preventing renal disease progression after AKI.
Keywords: CKD; acute renal failure; fibrosis; hypertension; hypoxia; tubular epithelium.
Copyright © 2015 by the American Society of Nephrology.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
