

Enzyme replacement and substrate reduction therapy for Gaucher disease
- PMID: 25812601
- PMCID: PMC8923052
- DOI: 10.1002/14651858.CD010324.pub2
Enzyme replacement and substrate reduction therapy for Gaucher disease
Abstract
Background: Gaucher disease, a rare disorder, is caused by inherited deficiency of the enzyme glucocerebrosidase. It is unique among the ultra-orphan disorders in that four treatments are currently approved by various regulatory authorities for use in routine clinical practice. Hitherto, because of the relatively few people affected worldwide, many of whom started therapy during a prolonged period when there were essentially no alternatives to imiglucerase, these treatments have not been systematically evaluated in studies such as randomized controlled trials now considered necessary to generate the highest level of clinical evidence.
Objectives: To summarize all available randomized controlled study data on the efficacy and safety of enzyme replacement therapies and substrate reduction therapy for treating Gaucher disease.
Search methods: We searched the Cochrane Cystic Fibrosis and Genetic Disorders Group's Inborn Errors of Metabolism Trials Register. Additional searches were conducted on ClinicalTrials.gov for any ongoing studies with potential interim results, and through PubMed. We also searched the reference lists of relevant articles and reviews.Date of last search: 07 August 2014.
Selection criteria: All randomized and quasi-randomized controlled studies (including open-label studies and cross-over studies) assessing enzyme replacement therapy or substrate reduction therapy, or both, in all types of Gaucher disease were included.
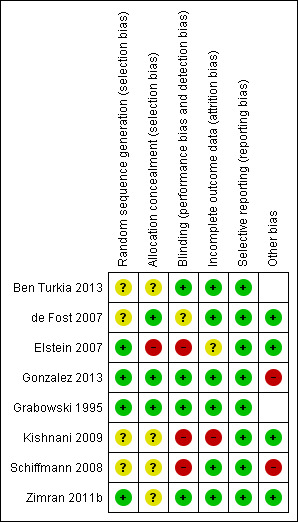
Data collection and analysis: Two authors independently assessed the risk of bias in the included studies, and extracted relevant data.
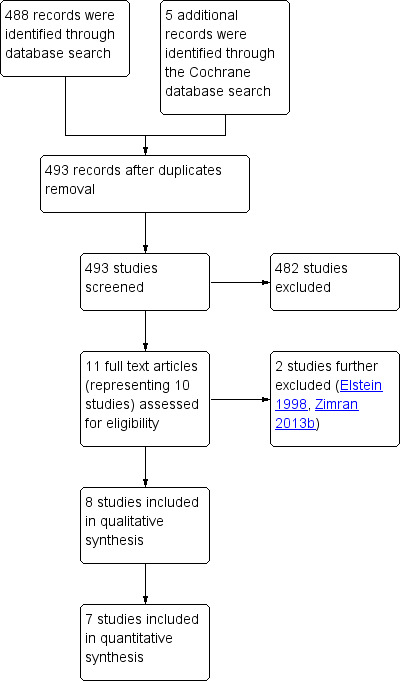
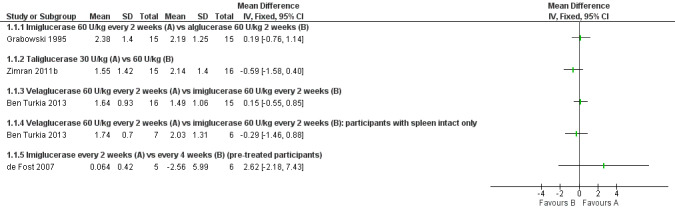
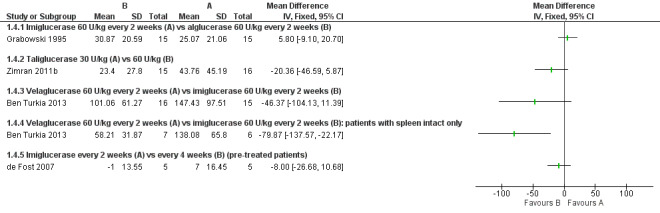
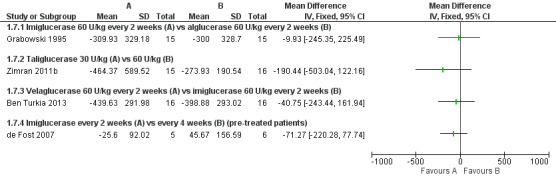
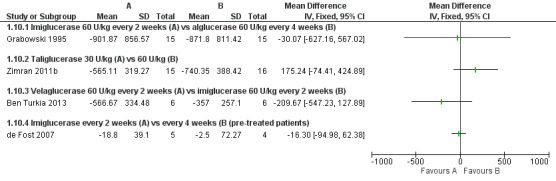
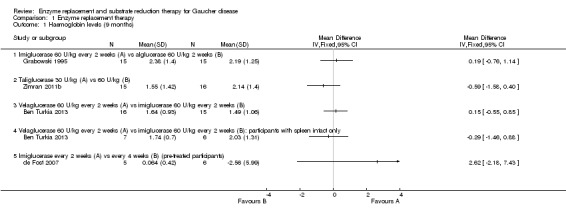
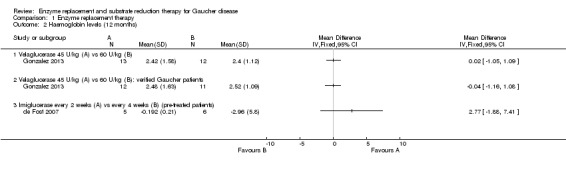
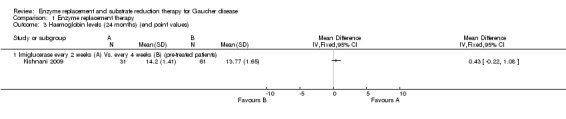
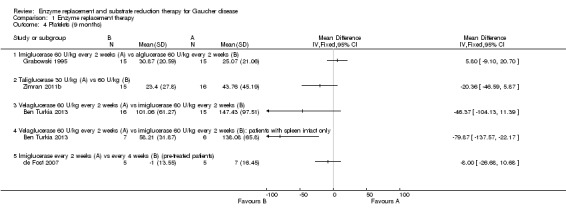
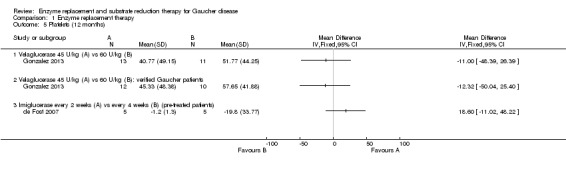
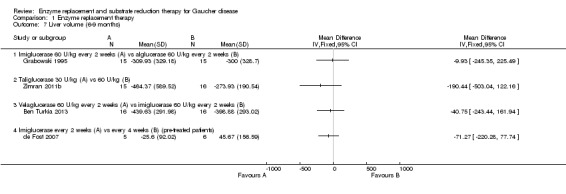
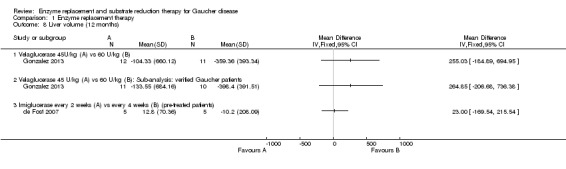
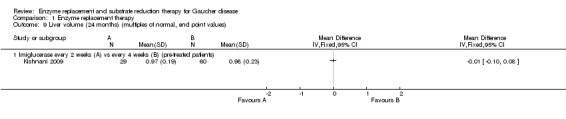
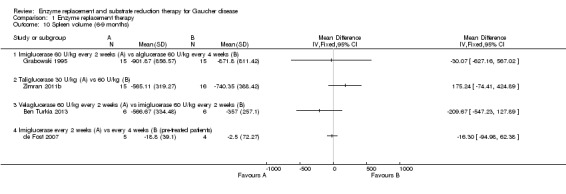
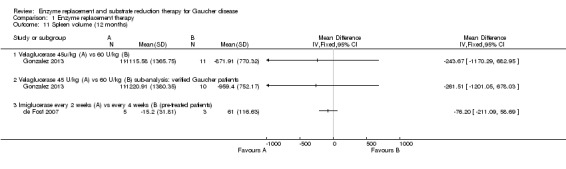
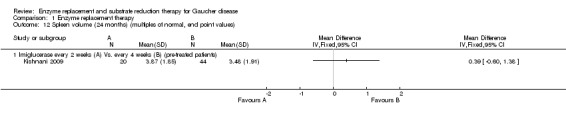
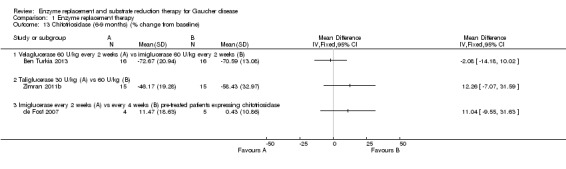
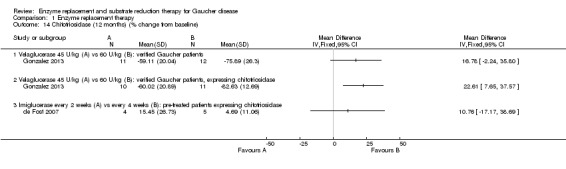
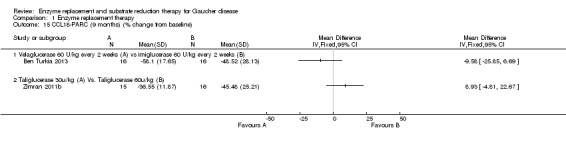
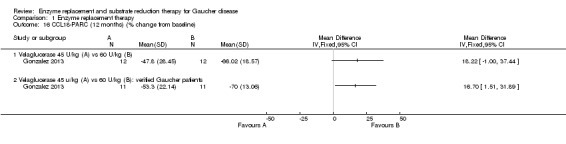
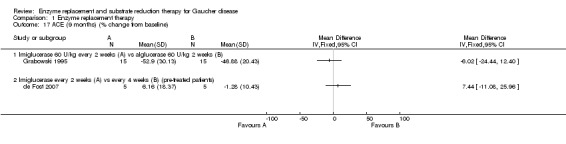
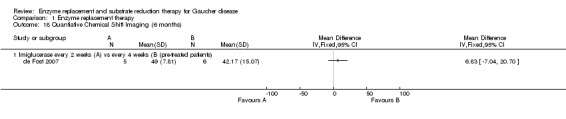
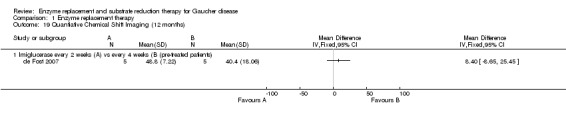
Main results: Of the 488 studies retrieved by the electronic searches, eight met the inclusion criteria and were analysed (300 participants). Response parameters were restricted to haemoglobin concentration, platelet count, spleen and liver volume and serum biomarkers (chitotriosidase and CCL18). Only one publication reported a 'low risk of bias' score in all parameters assessed, and all studies included were randomized.Four studies reported the responses to enzyme replacement therapy of previously untreated individuals with type 1 Gaucher disease. Two studies investigated maintenance enzyme replacement therapy in people with stable type 1 Gaucher disease previously treated for at least two years. One study compared substrate reduction therapy, enzyme replacement therapy and a combination thereof as maintenance therapy in people with type 1 Gaucher disease previously treated with enzyme replacement therapy. One study examined substrate reduction therapy in people with chronic neuronopathic (type 3) Gaucher disease who continued to receive enzyme replacement therapy.Treatment-naïve participants had similar increases in haemoglobin when comparing those receiving imiglucerase or alglucerase at 60 units/kg, imiglucerase or velaglucerase alfa at 60 U/kg, taliglucerase alfa at 30 units/kg or 60 units/kg, and velaglucerase alfa at 45 units/g or 60 units/kg. For platelet count response in participants with intact spleens, a benefit for imiglucerase over velaglucerase alfa at 60 units/kg was observed, mean difference -79.87 (95% confidence interval -137.57 to -22.17). There were no other significant differences in platelet count response when comparing different doses of velaglucerase alfa and of taliglucerase alfa, and when comparing imiglucerase to alglucerase. Spleen and liver volume reductions were not significantly different in any enzyme replacement therapy product or dose comparison study. Although a dose effect on serum biomarkers was not seen after nine months, a significantly greater reduction with higher dose was reported after 12 months in the velaglucerase study, mean difference 16.70 (95% confidence intervaI 1.51 to 31.89). In the two enzyme replacement therapy maintenance studies comparing infusions every two weeks and every four weeks, there were no significant differences in haemoglobin concentration, platelet count, and spleen and liver volumes over a 6 to 12 month period when participants were treated with the same cumulative dose.A total of 25 serious adverse events were reported, nearly all deemed unrelated to treatment.There are, as yet, no randomized trials of substrate reduction therapy in treatment-naïve patients that can be evaluated. Miglustat monotherapy appeared as effective as continued enzyme replacement therapy for maintenance of hematological, organ and biomarker responses in people with type 1 Gaucher disease previously treated with imiglucerase for at least two years. In those with neuronopathic Gaucher disease, no significant improvements in haemoglobin concentration, platelet count or organ volumes occurred when enzyme replacement therapy was augmented with miglustat.One randomized controlled study assessing substrate reduction therapy was published immediately prior to producing the final version of this review, and this, along with a further ongoing study (expected to be published in the near future), will be assessed for eligibility in a future update of the review.
Authors' conclusions: The results reflect the limitations of analysing evidence restricted to prospective randomized controlled trials, especially when dealing with chronic rare diseases. This analysis suggests that, during the first year of treatment, different recombinant glucocerebrosidases are bio-similar and non-inferior in safety and efficacy for surrogate biological response parameters. Enzyme replacement therapy given at 30 to 45 units/kg body weight every two to four weeks was generally as effective as the 60 unit/kg dose for the assessed clinical outcomes. The analysis emphasise the need to determine whether it is realistic to carry out multi-decade prospective clinical trials for rare diseases such as type 1 Gaucher disease. With large treatment effects on the classical manifestations of the disorder, therapeutic investigations in Gaucher disease mandate innovative trial designs and methodology to secure decisive data concerning long-term efficacy and safety - with the realization that knowledge about disease-modifying actions that are sustained are of crucial importance to people with this chronic condition.
Conflict of interest statement
Dr Elad Shemesh: none known.
Dr Laura Deroma: no direct conflicts of interest but her Institution received some equipment and consumables from pharmaceutical companies (Genzyme, Shire, Actelion). These also co‐financed several research projects.
Dr Bruno Bembi: a member of a bio‐tech university spin‐off (University of Udine) named Transactiva, operating in the field of vegetal bio reactor protein production. His institution received from pharmaceutical companies (Shire, Genzyme, Actelion) grants and co‐financing (equipments and consumables) for research projects; the author participated to meetings/advisory boards promoted by these companies. He is member of a bio‐tech university spin‐off named Transactiva, operating in the field of vegetal bio reacter protein production.
Dr Patrick Deegan: declares no bias toward any company mentioned or its products.
Prof Carla Hollak: received fees and travel reimbursements for consultancies and/or lectures in the field of lysosomal storage disorders from Genzyme, Shire, Protalix and Actelion. She has received grants for educational purposes and for a public‐private partnership TIPharma project, from Genzyme and Shire. All fees are received by the AMC Medical research BV in accordance with AMC regulations.
Dr Neal Weinreb: received research support (Genzyme/Sanofi, Shire HGT) and honoraria (Genzyme/Sanofi, Shire HGT,Pfizer) and has served on advisory boards (Genzyme/Sanofi, Shire HGT, Pfizer, Protalix Biotherapeutics) and speakers bureaus (Genzyme/Sanofi, Actelion, Pfizer).
Professor Timothy Cox: interests in this clinical and scientific field are longstanding (over 25 years) and, where possible, he endeavours to work with all companies in this ultra‐orphan field to promote competitive clinical and laboratory science for the benefit of patients with Gaucher disease; his personal rewards are relatively modest.
Figures

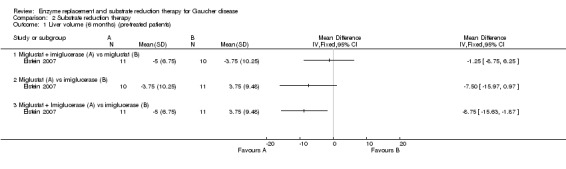
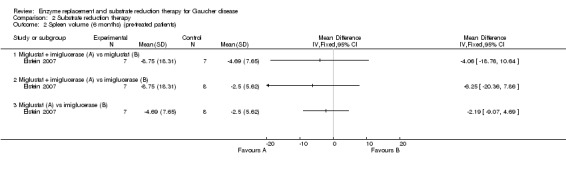
Update of
- doi: 10.1002/14651858.CD010324
Similar articles
-
Sertindole for schizophrenia.Cochrane Database Syst Rev. 2005 Jul 20;2005(3):CD001715. doi: 10.1002/14651858.CD001715.pub2. Cochrane Database Syst Rev. 2005. PMID: 16034864 Free PMC article.
-
Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis.Cochrane Database Syst Rev. 2021 Apr 19;4(4):CD011535. doi: 10.1002/14651858.CD011535.pub4. Cochrane Database Syst Rev. 2021. Update in: Cochrane Database Syst Rev. 2022 May 23;5:CD011535. doi: 10.1002/14651858.CD011535.pub5. PMID: 33871055 Free PMC article. Updated.
-
Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis.Cochrane Database Syst Rev. 2017 Dec 22;12(12):CD011535. doi: 10.1002/14651858.CD011535.pub2. Cochrane Database Syst Rev. 2017. Update in: Cochrane Database Syst Rev. 2020 Jan 9;1:CD011535. doi: 10.1002/14651858.CD011535.pub3. PMID: 29271481 Free PMC article. Updated.
-
Enzyme replacement therapy for infantile-onset Pompe disease.Cochrane Database Syst Rev. 2017 Nov 20;11(11):CD011539. doi: 10.1002/14651858.CD011539.pub2. Cochrane Database Syst Rev. 2017. PMID: 29155436 Free PMC article.
-
Drugs for preventing postoperative nausea and vomiting in adults after general anaesthesia: a network meta-analysis.Cochrane Database Syst Rev. 2020 Oct 19;10(10):CD012859. doi: 10.1002/14651858.CD012859.pub2. Cochrane Database Syst Rev. 2020. PMID: 33075160 Free PMC article.
Cited by
-
Successful switch from enzyme replacement therapy to miglustat in an adult patient with type 1 Gaucher disease: a case report.J Med Case Rep. 2016 Nov 8;10(1):315. doi: 10.1186/s13256-016-1060-y. J Med Case Rep. 2016. PMID: 27821156 Free PMC article.
-
Accuracy of chitotriosidase activity and CCL18 concentration in assessing type I Gaucher disease severity. A systematic review with meta-analysis of individual participant data.Haematologica. 2021 Feb 1;106(2):437-445. doi: 10.3324/haematol.2019.236083. Haematologica. 2021. PMID: 32001533 Free PMC article.
-
Lysosomal storage diseases.Transl Sci Rare Dis. 2017 May 25;2(1-2):1-71. doi: 10.3233/TRD-160005. Transl Sci Rare Dis. 2017. PMID: 29152458 Free PMC article. Review.
-
Natural history of inflammation and impaired autophagy in children with Gaucher disease identified by newborn screening.Mol Genet Metab Rep. 2025 Jan 21;42:101187. doi: 10.1016/j.ymgmr.2025.101187. eCollection 2025 Mar. Mol Genet Metab Rep. 2025. PMID: 39902270 Free PMC article.
-
Home Enzyme Replacement Therapy in Gaucher Disease: A Review.J Clin Med. 2025 Jan 27;14(3):842. doi: 10.3390/jcm14030842. J Clin Med. 2025. PMID: 39941513 Free PMC article. Review.
References
References to studies included in this review
Ben Turkia 2013 {published data only}
-
- Ben Turkia H, Gonzalez DE, Barton NW, Zimran A, Kabra M, Lukina EA, et al. Velaglucerase alfa enzyme replacement therapy compared with imiglucerase in patients with Gaucher disease. American Journal of Hematology 2013;88(3):179‐84. [PUBMED: 23400823] - PubMed
-
- Ben Turkia H, Gonzalez DE, Kabra M, Lukina EA, Giraldo P, Kisinovsky I, et al. Two‐year safety and tolerability of velaglucerase alfa in patients with type 1 gaucher disease, including patients switched from imiglucerase: phase III trial HGT‐GCB‐039 and extension [abstract]. Blood 2011;118(21):Poster: 3214. [CENTRAL: 977632; CRS: 5500125000000576]
-
- Ben Turkia H, Gonzalez DE, Zimran A, Kabra M, Lukina EA, Giraldo P, et al. Achievement of therapeutic goals in patients with type 1 gaucher disease (GD1) on velaglucerase alfa or imiglucerase: phase III trials HGT‐GCB‐039 and extension [abstract]. Journal of Inherited Metabolic Disease 2011;34 Suppl 3:S224, Abstract no: P‐432. [CENTRAL: 977631; CRS: 5500125000000575]
-
- Elstein D, Ben Turkia H, Gonzalez DE, Kabra M, Lukina EA, Giraldo P, et al. Bone mineral density in adults with type 1 gaucher disease receiving velaglucerase alfa 60 U/KG every other week: 2‐year results [abstract]. Journal of Inherited Metabolic Disease 2012;35 Suppl 1:S150, Abstract no: P‐410. [CENTRAL: 977634; CRS: 5500125000000578]
-
- Zimran A, Kisinovsky I, Lukina EA, Elstein D, Zahrieh D, Crombez E, et al. Efficacy of long‐term velaglucerase alfa on haematological and visceral parameters in patients with type 1 gaucher disease [abstract]. Journal of Inherited Metabolic Disease 2013;36 Suppl 2:S302, Abstract no: P‐728. [CENTRAL: 977639; CRS: 5500125000000583]
de Fost 2007 {published data only}
-
- Fost M, Aerts JM, Groener JE, Maas M, Akkerman EM, Wiersma MG, et al. Low frequency maintenance therapy with imiglucerase in adult type I Gaucher disease: a prospective randomized controlled trial. Haematologica 2007;92(2):215‐21. [PUBMED: 17296571] - PubMed
Elstein 2007 {published data only}
-
- Elstein D, Dweck A, Attias D, Hadas‐Halpern I, Zevin S, Altarescu G, et al. Oral maintenance clinical trial with miglustat for type I Gaucher disease: switch from or combination with intravenous enzyme replacement. Blood 2007;110(7):2296‐301. [PUBMED: 17609429] - PubMed
-
- Elstein D, Heitner R, Dweck A, Attias D, Atarescu G, Zimran A. OGT 918 as substrate reduction therapy in type I gaucher disease [abstract]. 6th Meeting of the European Haematology Association; 2001 June 21‐24; Frankfurt, Germany.. 2001:Abstract no: 212. [CENTRAL: 593107; CRS: 5500100000003060]
Gonzalez 2013 {published data only}
-
- Elstein D, Ben Turkia H, Gonzalez DE, Kabra M, Lukina EA, Giraldo P, et al. Bone mineral density in adults with type 1 gaucher disease receiving velaglucerase alfa 60 U/KG every other week: 2‐year results [abstract]. Journal of Inherited Metabolic Disease 2012;35 Suppl 1:S150, Abstract no: P‐410. [CENTRAL: 977634; CRS: 5500125000000578]
-
- Gonzalez DE, Ben Dridi MF, Lukina E, Kisinovsky I, Ben Turkia H, Elstein D, et al. Clinically significant hemoglobin response observed within 3 months following treatment with velaglucerase alfa in patients with type 1 gaucher disease [abstract]. Journal of Inherited Metabolic Disease 2010;33 Suppl 1:S139, Abstract no: 442‐P. [CENTRAL: 977630; CRS: 5500125000000574]
-
- Gonzalez DE, Turkia HB, Lukina EA, Kisinovsky I, Dridi MF, Elstein D, et al. Enzyme replacement therapy with velaglucerase alfa in Gaucher disease: Results from a randomized, double‐blind, multinational, Phase 3 study. American Journal of Hematology 2013;88(3):166‐71. [PUBMED: 23386328] - PubMed
-
- Zimran A, Kisinovsky I, Lukina EA, Elstein D, Zahrieh D, Crombez E, et al. Efficacy of long‐term velaglucerase alfa on haematological and visceral parameters in patients with type 1 gaucher disease [abstract]. Journal of Inherited Metabolic Disease 2013;36 Suppl 2:S302, Abstract no: P‐728. [CENTRAL: 977639; CRS: 5500125000000583]
Grabowski 1995 {published data only}
-
- Grabowski GA, Barton NW, Pastores G, Dambrosia JM, Banerjee TK, McKee MA, et al. Enzyme therapy in type 1 Gaucher disease: comparative efficacy of mannose‐terminated glucocerebrosidase from natural and recombinant sources. Annals of Internal Medicine 1995;122(1):33‐9. [PUBMED: 7985893] - PubMed
-
- Grabowski GA, Barton NW, Pastores G, Dambrosia JM, Banerjee TK, McKee MA, et al. Enzyme therapy in gaucher disease type 1: comparative efficacy of mannose‐terminated glucocerebrosidase from natural and recombinant sources [abstract]. Blood 1994, issue Suppl:226a. - PubMed
Kishnani 2009 {published data only}
-
- Kishnani PS, DiRocco M, Kaplan P, Mehta A, Pastores GM, Smith SE, et al. A randomized trial comparing the efficacy and safety of imiglucerase (Cerezyme) infusions every 4 weeks versus every 2 weeks in the maintenance therapy of adult patients with Gaucher disease type 1. Molecular Genetics and Metabolism 2009;96(4):164‐70. [PUBMED: 19195916] - PubMed
Schiffmann 2008 {published data only}
Zimran 2011b {published data only}
-
- Zimran A, Brill‐Almon E, Chertkoff R, Petakov M, Blanco‐Favela F, Munoz ET, et al. Pivotal trial with plant cell‐expressed recombinant glucocerebrosidase, taliglucerase alfa, a novel enzyme replacement therapy for Gaucher disease. Blood 2011;118(22):5767‐73. [PUBMED: 21900191] - PubMed
-
- Zimran A, Gonzalez‐Rodriguez DE, Abrahamov A, Elstein D, Heitner R, Paz A, et al. A multicenter, double‐blind, randomized safety and efficacy study of two dose levels of taliglucerase alfa in pediatric patients with gaucher disease [abstract]. Blood 2012;120(21):Abstract no: 2140. [CENTRAL: 977633; CRS: 5500125000000577]
-
- Zimran A, Gonzalez‐Rodriguez DE, Abrahamov A, Elstein D, Paz A, Brill‐Almon E, et al. A multi‐center, double‐blind, randomized safety and efficacy study of two dose levels of taliglucerase alfa in pediatric patients with gaucher disease [abstract]. Journal of Inherited Metabolic Disease 2013;36 Suppl 2:S301, Abstract no: P‐724. [CENTRAL: 977638; CRS: 5500125000000582]
-
- Zimran A, Mehta A, Giraldo P, Rosenbaum H, Giona F, Amato D, et al. Long‐term safety and efficacy data of taliglucerase alfa, a plant cell expressed recombinant glucocerebrosidase, in the treatment of naive gaucher disease patients [abstract]. Journal of Inherited Metabolic Disease 2012;35 Suppl 1:S11, Abstract no: O‐032. [CENTRAL: 977635; CRS: 5500125000000579]
-
- Dussen L, Zimran A, Akkerman EM, Aerts JM, Petakov M, Elstein D, et al. Taliglucerase alfa leads to favorable bone marrow responses in patients with type I Gaucher disease. Blood cells, Molecules & Diseases 2013;50(3):206‐11. [PUBMED: 23199589] - PubMed
References to studies excluded from this review
Elstein 1998 {published data only}
-
- Elstein D, Abrahamov A, Hadas‐Halpern I, Meyer A, Zimran A. Low‐dose low‐frequency imiglucerase as a starting regimen of enzyme replacement therapy for patients with type I Gaucher disease. QJM: Monthly Journal of the Association of Physicians 1998;91(7):483‐8. [PUBMED: 9797931] - PubMed
Wenstrup 2004 {published data only}
-
- Wenstrup RJ, Bailey L, Grabowski GA, Moskovitz J, Oestreich AE, Wu W, et al. Gaucher disease: alendronate disodium improves bone mineral density in adults receiving enzyme therapy. Blood 2004;104(5):1253‐7. [CENTRAL: 489109; CRS: 5500100000002632; PUBMED: 15010365] - PubMed
Zimran 2013b {published data only}
References to studies awaiting assessment
Engage 2015 {published data only}
-
- Ben Turkia HMF, Lukina E, Amato D, Baris H, Dasouki M, Ghosn M, et al. Engage: a phase 3, randomized, double blind, placebo controlled, multi center study to investigate the efficacy and safety of eliglustat in adults with gaucher disease type 1: 9 month results [abstract]. Journal of Inherited Metabolic Disease 2013;36 Suppl 2:S268, Abstract no: P‐596. [CENTRAL: 977640; CRS: 5500125000000584]
-
- Dasouka M, Lukina E, Ben Dridi MF, Amato D, Baris H, Ghosn M, et al. Effects of oral eliglustat on bone disease in gaucher disease type 1: results from the randomized, placebo‐controlled engage trial [abstract]. Journal of Inherited Metabolic Disease 2013;36 Suppl 2:S268, Abstract no: P‐597. [CENTRAL: 977636; CRS: 5500125000000580]
References to ongoing studies
Encore 2013 {published data only}
-
- Cox TM, Drelichman G, Cravo R, Balwani M, Burrow T, Martins AM, et al. Encore: a multi‐national, randomized, controlled, open‐label non‐inferiority study comparing eliglustat to imiglucerase in gaucher disease type 1 (GD1) patients on enzyme replacement therapy (ERT) who have reached therapeutic goals [abstract]. Journal of Inherited Metabolic Disease 2013;36 Suppl 2:S268, Abstract no: P‐598. [CENTRAL: 977637; CRS: 5500125000000581]
Additional references
Aerts 2006
-
- Aerts JM, Hollak CE, Boot RG, Groener JE, Maas M. Substrate reduction therapy of glycosphingolipid storage disorders. Journal of Inherited Metabolic Disease 2006;29(2‐3):449‐56. [PUBMED: 16763917] - PubMed
Amiri 2012
-
- Amiri M, Naim HY. Miglustat‐induced intestinal carbohydrate malabsorption is due to the inhibition of alpha‐glucosidases, but not beta‐galactosidases. Journal of Inherited Metabolic Disease 2012;35(6):949‐54. [PUBMED: 22976762] - PubMed
Andersson 2008
-
- Andersson H, Kaplan P, Kacena K, Yee J. Eight‐year clinical outcomes of long‐term enzyme replacement therapy for 884 children with Gaucher disease type 1. Pediatrics 2008;122(6):1182‐90. [PUBMED: 19047232] - PubMed
Barton 1991
-
- Barton NW, Brady RO, Dambrosia JM, Bisceglie AM, Doppelt SH, Hill SC, et al. Replacement therapy for inherited enzyme deficiency‐‐macrophage‐targeted glucocerebrosidase for Gaucher's disease. New England Journal of Medicine 1991;324(21):1464‐70. [PUBMED: 2023606] - PubMed
Beutler 1993
Beutler 2006
-
- Beutler E. Gaucher disease: multiple lessons from a single gene disorder. Acta Paediatrica (Oslo, Norway : 1992). Supplement 2006;95(451):103‐9. [PUBMED: 16720474] - PubMed
Biegstraaten 2010
-
- Biegstraaten M, Mengel E, Marodi L, Petakov M, Niederau C, Giraldo P, et al. Peripheral neuropathy in adult type 1 Gaucher disease: a 2‐year prospective observational study. Brain: a Journal of Neurology 2010;133(10):2909‐19. [PUBMED: 20693542] - PubMed
Black 1996
Brady 1966
Bultron 2010
Charrow 2000
-
- Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G, et al. The Gaucher registry: demographics and disease characteristics of 1698 patients with Gaucher disease. Archives of Internal Medicine 2000;160(18):2835‐43. [PUBMED: 11025794] - PubMed
Cherin 2006
-
- Cherin P, Sedel F, Mignot C, Schupbach M, Gourfinkel‐An I, Verny M, et al. Neurological manifestations of type 1 Gaucher's disease: Is a revision of disease classification needed? [Les manifestations neurologiques de la maladie de Gaucher de type 1: vers une remise en cause de la classification actuelle?]. Revue Neurologique 2006;162(11):1076‐83. [PUBMED: 17086144] - PubMed
Chetrit 2013
-
- Chetrit EB, Alcalay RN, Steiner‐Birmanns B, Altarescu G, Phillips M, Elstein D, et al. Phenotype in patients with Gaucher disease and Parkinson disease. Blood Cells, Molecules & Diseases 2013;50(3):218‐21. [PUBMED: 23265741] - PubMed
Cole 2011
Conradi 1991
-
- Conradi N, Kyllerman M, Mansson JE, Percy AK, Svennerholm L. Late‐infantile Gaucher disease in a child with myoclonus and bulbar signs: neuropathological and neurochemical findings. Acta Neuropathologica 1991;82(2):152‐7. [PUBMED: 1718128] - PubMed
Cox 2000
-
- Cox T, Lachmann R, Hollak C, Aerts J, Weely S, Hrebicek M, et al. Novel oral treatment of Gaucher's disease with N‐butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet 2000;355(9214):1481‐5. [PUBMED: 10801168] - PubMed
Cox 2003
-
- Cox TM, Aerts JM, Andria G, Beck M, Belmatoug N, Bembi B, et al. The role of the iminosugar N‐butyldeoxynojirimycin (miglustat) in the management of type I (non‐neuronopathic) Gaucher disease: a position statement. Journal of Inherited Metabolic Disease 2003;26(6):513‐26. [PUBMED: 14605497] - PubMed
Cox 2010
-
- Cox TM. Eliglustat tartrate, an orally active glucocerebroside synthase inhibitor for the potential treatment of Gaucher disease and other lysosomal storage diseases. Current Opinion in Investigational Drugs 2010;11(10):1169‐81. [PUBMED: 20872320] - PubMed
Damiano 1998
-
- Damiano AM, Pastores GM, Ware JE Jr. The health‐related quality of life of adults with Gaucher's disease receiving enzyme replacement therapy: results from a retrospective study. Quality of Life Research : an International Journal of Quality of Life Aspects of Treatment, Care and Rehabilitation 1998;7(5):373‐86. [PUBMED: 9691718] - PubMed
de Fost 2006
-
- Fost M, Hollak CE, Groener JE, Aerts JM, Maas M, Poll LW, et al. Superior effects of high‐dose enzyme replacement therapy in type 1 Gaucher disease on bone marrow involvement and chitotriosidase levels: a 2‐center retrospective analysis. Blood 2006;108(3):830‐5. [PUBMED: 16527890] - PubMed
Deeks 2011
-
- Deeks J, Higgins J, Altman D. Chapter 9 Analysing data and undertaking meta‐analysis. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Deroma 2013
Dietz 2010
-
- Dietz HC. New therapeutic approaches to mendelian disorders. The New England journal of medicine 2010;363(9):852‐63. [PUBMED: 20818846] - PubMed
Elbourne 2002
-
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. - PubMed
Genzyme/Sanofi 2013 [pers comm]
-
- Genzyme/Sanofi. Prescribed ERT infusions [personal communication]. Email to: Neal Weinreb 2013.
Gerstein 2012
-
- Gerstein HC, Bosch J, Dagenais GR, Díaz R, Jung H, Maggioni AP, et al. Basal insulin and cardiovascular and other outcomes in dysglycemia. New England Journal of Medicine 2012;367(4):319‐28. - PubMed
Giraldo 2011
-
- Giraldo P, Irun P, Alfonso P, Dalmau J, Fernandez‐Galan MA, Figueredo A, et al. Evaluation of Spanish Gaucher disease patients after a 6‐month imiglucerase shortage. Blood Cells, Molecules & Diseases 2011;46(1):115‐8. [PUBMED: 20934891] - PubMed
Goker‐Alpan 2004
Goldblatt 2011
-
- Goldblatt J, Fletcher JM, McGill J, Szer J, Wilson M. Enzyme replacement therapy "drug holiday": results from an unexpected shortage of an orphan drug supply in Australia. Blood cells, Molecules & Diseases 2011;46(1):107‐10. [PUBMED: 20684886] - PubMed
Grabowski 1997
-
- Grabowski GA. Gaucher disease: gene frequencies and genotype/phenotype correlations. Genetic testing 1997;1(1):5‐12. [PUBMED: 10464619] - PubMed
Grabowski 2008
-
- Grabowski GA. Phenotype, diagnosis, and treatment of Gaucher's disease. Lancet 2008;372(9645):1263‐71. - PubMed
Grabowski 2009
-
- Grabowski GA, Kacena K, Cole JA, Hollak CE, Zhang L, Yee J, et al. Dose‐response relationships for enzyme replacement therapy with imiglucerase/alglucerase in patients with Gaucher disease type 1. Genetics in Medicine: Official Journal of the American College of Medical Genetics 2009;11(2):92‐100. [PUBMED: 19265748] - PMC - PubMed
Higgins 2011a
-
- Higgins JPT, Altman DG (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hollak 2010
-
- Hollak CE, vom Dahl S, Aerts JM, Belmatoug N, Bembi B, Cohen Y, et al. Force majeure: therapeutic measures in response to restricted supply of imiglucerase (Cerezyme) for patients with Gaucher disease. Blood Cells, Molecules & Diseases 2010;44(1):41‐7. [PUBMED: 19804996] - PubMed
Hollak 2011
Hollak 2012
Hughes 2007
-
- Hughes DA, Mlilligan A, Mehta A. Home therapy for lysosomal storage disorders. British Journal of Nursing 2007;16(22):1386‐9. - PubMed
Inokuchi 1987
-
- Inokuchi J, Radin NS. Preparation of the active isomer of 1‐phenyl‐2‐decanoylamino‐3‐morpholino‐1‐propanol, inhibitor of murine glucocerebroside synthetase. Journal of Lipid Research 1987;28(5):565‐71. [PUBMED: 2955067] - PubMed
Kaplan 2006
-
- Kaplan P, Andersson HC, Kacena KA, Yee JD. The clinical and demographic characteristics of nonneuronopathic Gaucher disease in 887 children at diagnosis. Archives of Pediatrics & Adolescent Medicine 2006;160(6):603‐8. [PUBMED: 16754822] - PubMed
Kauli 2000
-
- Kauli R, Zaizov R, Lazar L, Pertzelan A, Laron Z, Galatzer A, et al. Delayed growth and puberty in patients with Gaucher disease type 1: natural history and effect of splenectomy and/or enzyme replacement therapy. Israel Medical Association Journal 2000;2(2):158‐63. [PUBMED: 10804944] - PubMed
Khan 2012
-
- Khan A, Hangartner T, Weinreb NJ, Taylor JS, Mistry PK. Risk factors for fractures and avascular osteonecrosis in type 1 Gaucher disease: a study from the International Collaborative Gaucher Group (ICGG) Gaucher Registry. Journal of Bone and Mineral Research : the Official Journal of the American Society for Bone and Mineral Research 2012;27(8):1839‐48. [PUBMED: 22692814] - PubMed
Krumholz 2013
Kuter 2013
-
- Kuter DJ, Mehta A, Hollak CE, Giraldo P, Hughes D, Belmatoug N, et al. Miglustat therapy in type 1 Gaucher disease: clinical and safety outcomes in a multicenter retrospective cohort study. Blood Cells, Molecules & Diseases 2013;51(2):116‐24. [PUBMED: 23683771] - PubMed
Langeveld 2008
-
- Langeveld M, Fost M, Aerts JM, Sauerwein HP, Hollak CE. Overweight, insulin resistance and type II diabetes in type I Gaucher disease patients in relation to enzyme replacement therapy. Blood Cells, Molecules & Diseases 2008;40(3):428‐32. [PUBMED: 17950007] - PubMed
Lee 1982
-
- Lee RE. The pathology of Gaucher disease. Progress in Clinical and Biological Research 1982;95:177‐217. [PUBMED: 7122634] - PubMed
Lukina 2010
-
- Lukina E, Watman N, Arreguin EA, Dragosky M, Iastrebner M, Rosenbaum H, et al. Improvement in hematological, visceral, and skeletal manifestations of Gaucher disease type 1 with oral eliglustat tartrate (Genz‐112638) treatment: 2‐year results of a phase 2 study. Blood 2010;116(20):4095‐8. [PUBMED: 20713962] - PMC - PubMed
McEachern 2007
-
- McEachern KA, Fung J, Komarnitsky S, Siegel CS, Chuang WL, Hutto E, et al. A specific and potent inhibitor of glucosylceramide synthase for substrate inhibition therapy of Gaucher disease. Molecular Genetics and Metabolism 2007;91(3):259‐67. [PUBMED: 17509920] - PubMed
Meikle 1999
-
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA 1999;281(3):249‐54. [PUBMED: 9918480] - PubMed
Mistry 1996
-
- Mistry PK, Wraight EP, Cox TM. Therapeutic delivery of proteins to macrophages: implications for treatment of Gaucher's disease. Lancet 1996;348(9041):1555‐9. [PUBMED: 8950883] - PubMed
Mistry 2009
Mistry 2011
Nathan 2013
Oulaidi 2011
-
- Oulaidi F, Front‐Deschamps S, Gallienne E, Lesellier E, Ikeda K, Asano N, et al. Second‐generation iminoxylitol‐based pharmacological chaperones for the treatment of Gaucher disease. ChemMedChem 2011;6(2):353‐61. [PUBMED: 21275057] - PubMed
Pastores 2010
-
- Pastores GM. Recombinant glucocerebrosidase (imiglucerase) as a therapy for Gaucher disease. BioDrugs: Clinical Immunotherapeutics, Biopharmaceuticals and Gene Therapy 2010;24(1):41‐7. [PUBMED: 20055531] - PubMed
Platt 1994
-
- Platt FM, Neises GR, Dwek RA, Butters TD. N‐butyldeoxynojirimycin is a novel inhibitor of glycolipid biosynthesis. The Journal of Biological Chemistry 1994;269(11):8362‐5. [PUBMED: 8132559] - PubMed
Resnick 2013
-
- Resnick D, Bozic KJ. Meta‐analysis of Trials of Recombinant Human Bone Morphogenetic Protein‐2: What Should Spine Surgeons and Their Patients Do With This Information?. Annals of Internal Medicine 2013;158(12):912‐3. [PUBMED: 23778909] - PubMed
Rosenbloom 2005
-
- Rosenbloom BE, Weinreb NJ, Zimran A, Kacena KA, Charrow J, Ward E. Gaucher disease and cancer incidence: a study from the Gaucher Registry. Blood 2005;105(12):4569‐72. [PUBMED: 15718419] - PubMed
Rosenbloom 2011
Rosenbloom 2013
-
- Rosenbloom BE, Weinreb NJ. Gaucher disease: a comprehensive review. Critical Reviews in Oncogenesis 2013;18(3):163‐75. [PUBMED: 23510062] - PubMed
Sidransky 2004
-
- Sidransky E. Gaucher disease: complexity in a "simple" disorder. Molecular Genetics and Metabolism 2004;83(1‐2):6‐15. [PUBMED: 15464415] - PubMed
Starzyk 2007
-
- Starzyk K, Richards S, Yee J, Smith SE, Kingma W. The long‐term international safety experience of imiglucerase therapy for Gaucher disease. Molecular Genetics and Metabolism 2007;90(2):157‐63. [PUBMED: 17079176] - PubMed
Stein 2010
Stowens 1985
-
- Stowens DW, Teitelbaum SL, Kahn AJ, Barranger JA. Skeletal complications of Gaucher disease. Medicine 1985;64(5):310‐22. [PUBMED: 4033409] - PubMed
Weinreb 2002
-
- Weinreb NJ, Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, et al. Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. American Journal of Medicine 2002;113(2):112‐9. [PUBMED: 12133749] - PubMed
Zimran 2011a
-
- Zimran A, Altarescu G, Elstein D. Nonprecipitous changes upon withdrawal from imiglucerase for Gaucher disease because of a shortage in supply. Blood Cells, Molecules & Diseases 2011;46(1):111‐4. [PUBMED: 20542712] - PubMed
Zimran 2013a
-
- Zimran A, Altarescu G, Elstein D. Pilot study using ambroxol as a pharmacological chaperone in type 1 Gaucher disease. Blood cells, molecules & diseases 2013;50(2):134‐7. [PUBMED: 23085429] - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous