

SNPfisher: tools for probing genetic variation in laboratory-reared zebrafish
- PMID: 25813542
- PMCID: PMC4392598
- DOI: 10.1242/dev.118786
SNPfisher: tools for probing genetic variation in laboratory-reared zebrafish
Abstract
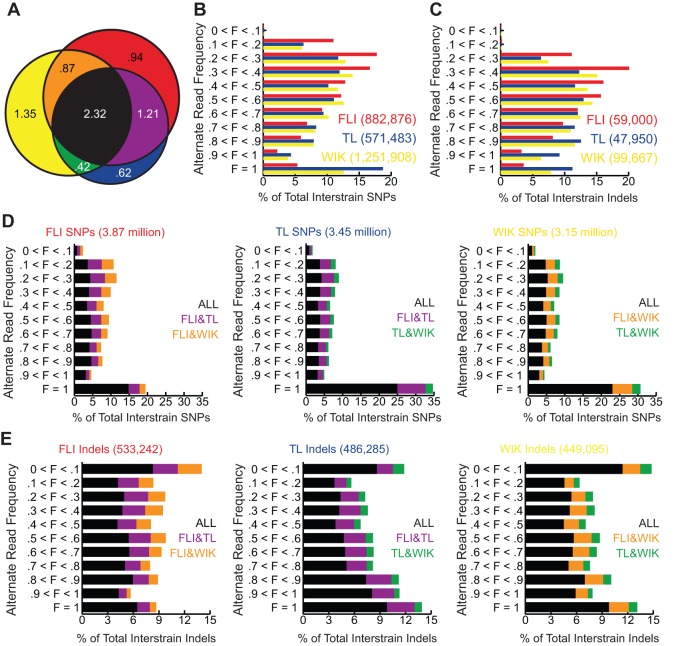
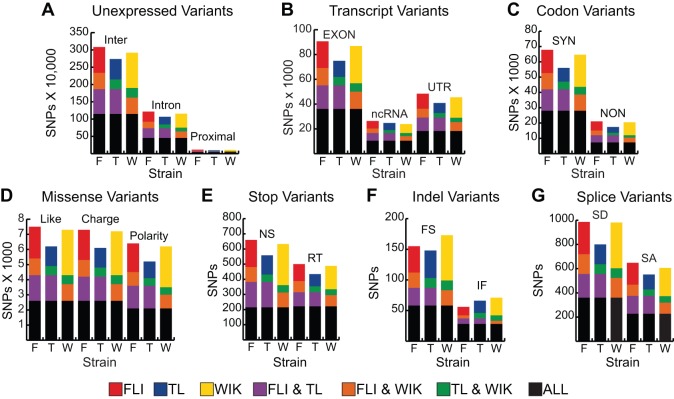
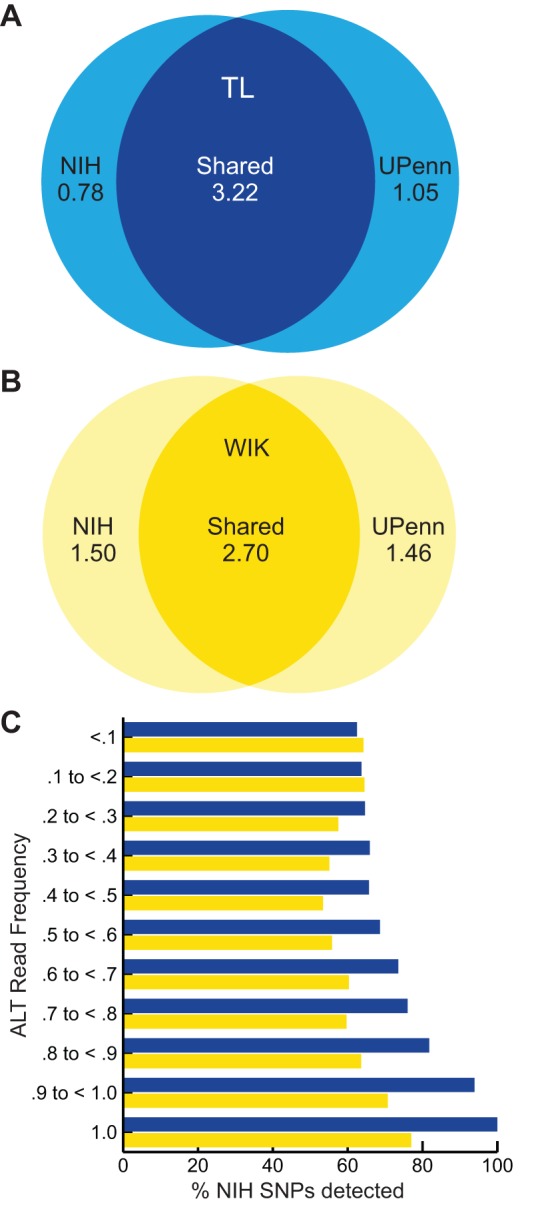
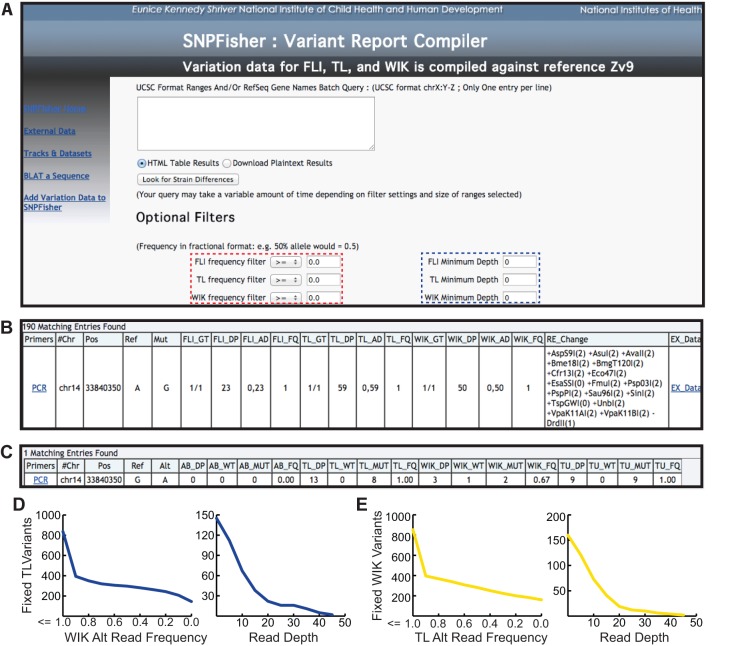
Single nucleotide polymorphisms (SNPs) are the benchmark molecular markers for modern genomics. Until recently, relatively few SNPs were known in the zebrafish genome. The use of next-generation sequencing for the positional cloning of zebrafish mutations has increased the number of known SNP positions dramatically. Still, the identified SNP variants remain under-utilized, owing to scant annotation of strain specificity and allele frequency. To address these limitations, we surveyed SNP variation in three common laboratory zebrafish strains using whole-genome sequencing. This survey identified an average of 5.04 million SNPs per strain compared with the Zv9 reference genome sequence. By comparing the three strains, 2.7 million variants were found to be strain specific, whereas the remaining variants were shared among all (2.3 million) or some of the strains. We also demonstrate the broad usefulness of our identified variants by validating most in independent populations of the same laboratory strains. We have made all of the identified SNPs accessible through 'SNPfisher', a searchable online database (snpfisher.nichd.nih.gov). The SNPfisher website includes the SNPfisher Variant Reporter tool, which provides the genomic position, alternate allele read frequency, strain specificity, restriction enzyme recognition site changes and flanking primers for all SNPs and Indels in a user-defined gene or region of the zebrafish genome. The SNPfisher site also contains links to display our SNP data in the UCSC genome browser. The SNPfisher tools will facilitate the use of SNP variation in zebrafish research as well as vertebrate genome evolution.
Keywords: Danio rerio; Genome; Next-generation sequencing; SNP; Variation; Whole-genome sequencing; Zebrafish.
© 2015. Published by The Company of Biologists Ltd.
Figures

References
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
