

Novel homozygous large deletion including the 5' part of the SPATA7 gene in a consanguineous Israeli Muslim Arab family
- PMID: 25814828
- PMCID: PMC4360169
Novel homozygous large deletion including the 5' part of the SPATA7 gene in a consanguineous Israeli Muslim Arab family
Abstract
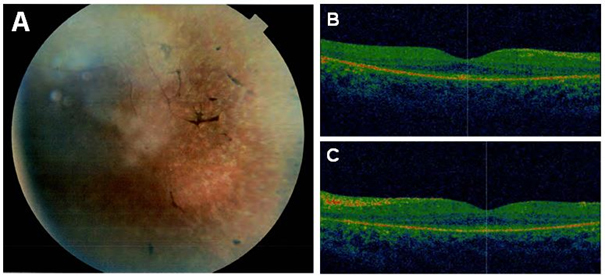
Purpose: To identify the genetic defect in a consanguineous Israeli Muslim Arab family with juvenile retinitis pigmentosa (RP).
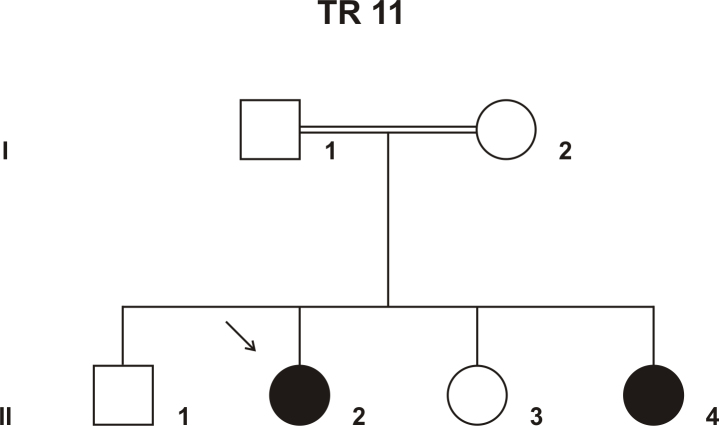
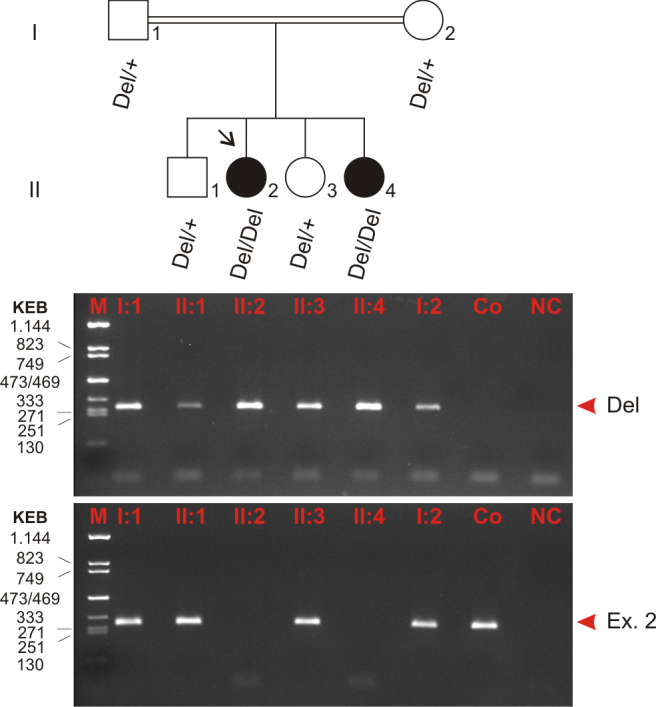
Methods: DNA samples were collected from the index patient, her parents, her affected sister, and two non-affected siblings. Genome-wide linkage analysis with 250 K single nucleotide polymorphism (SNP) arrays was performed using DNA from the two affected patients. Owing to consanguinity in the family, we applied homozygosity mapping to identify the disease-causing gene. The candidate gene SPATA7 was screened for mutations with PCR amplifications and direct Sanger sequencing.
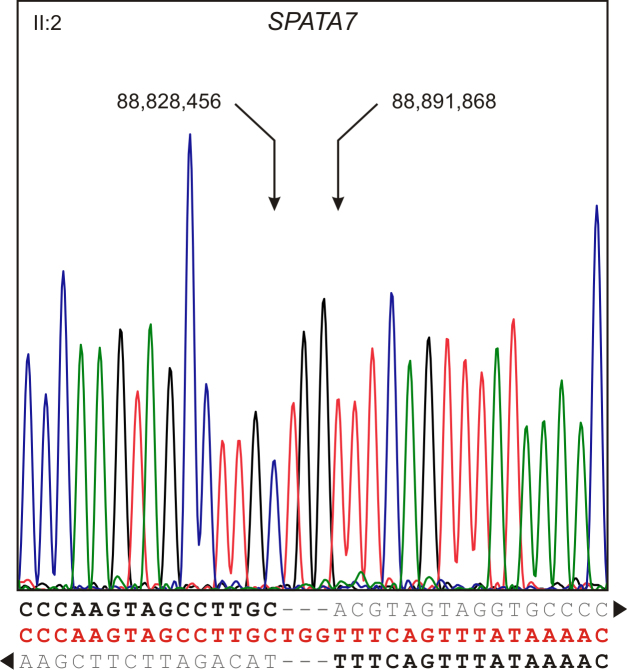
Results: Following high-density SNP arrays, we identified several homozygous genomic regions one of which included the SPATA7 gene. Several mutations in SPATA7 have been reported for various forms of retinal dystrophy, including Leber congenital amaurosis (LCA) and juvenile RP. PCR-based sequence content mapping, long-distance PCR amplifications, and subsequent sequencing analysis revealed a homozygous 63.4 kb large deletion that encompasses the 5' part of the SPATA7 gene including exons 1-5. The mutation showed concordant segregation with the phenotype in the family as expected for autosomal recessive mode of inheritance and is consistent with a diagnosis of juvenile RP.
Conclusions: We report a novel homozygous large deletion in SPATA7 associated with juvenile RP in a consanguineous Israeli Muslim Arab family. This is the first larger deletion mutation reported for SPATA7.
Figures
Similar articles
-
Identification of novel mutations in patients with Leber congenital amaurosis and juvenile RP by genome-wide homozygosity mapping with SNP microarrays.Invest Ophthalmol Vis Sci. 2007 Dec;48(12):5690-8. doi: 10.1167/iovs.07-0610. Invest Ophthalmol Vis Sci. 2007. PMID: 18055821
-
Identification of mutations causing inherited retinal degenerations in the israeli and palestinian populations using homozygosity mapping.Invest Ophthalmol Vis Sci. 2014 Feb 24;55(2):1149-60. doi: 10.1167/iovs.13-13625. Invest Ophthalmol Vis Sci. 2014. PMID: 24474277
-
A novel splice-site mutation of TULP1 underlies severe early-onset retinitis pigmentosa in a consanguineous Israeli Muslim Arab family.Mol Vis. 2008 Apr 21;14:675-82. Mol Vis. 2008. PMID: 18432314 Free PMC article.
-
A Novel Homozygous Mutation in SPTBN2 Leads to Spinocerebellar Ataxia in a Consanguineous Family: Report of a New Infantile-Onset Case and Brief Review of the Literature.Cerebellum. 2018 Jun;17(3):276-285. doi: 10.1007/s12311-017-0893-2. Cerebellum. 2018. PMID: 29196973 Review.
-
Discovery of rare homozygous mutations from studies of consanguineous pedigrees.Curr Protoc Hum Genet. 2012 Oct;Chapter 6:Unit6.12. doi: 10.1002/0471142905.hg0612s75. Curr Protoc Hum Genet. 2012. PMID: 23074070 Review.
Cited by
-
Unraveling the genetic cause of hereditary ophthalmic disorders in Arab societies from Israel and the Palestinian Authority.Eur J Hum Genet. 2020 Jun;28(6):742-753. doi: 10.1038/s41431-019-0566-3. Epub 2020 Jan 2. Eur J Hum Genet. 2020. PMID: 31896775 Free PMC article.
-
Spectrum, frequency, and genotype-phenotype of mutations in SPATA7.Mol Vis. 2019 Dec 2;25:821-833. eCollection 2019. Mol Vis. 2019. PMID: 31908400 Free PMC article.
-
Phenotypic expansion and progression of SPATA7-associated retinitis pigmentosa.Doc Ophthalmol. 2018 Apr;136(2):125-133. doi: 10.1007/s10633-018-9626-1. Epub 2018 Feb 6. Doc Ophthalmol. 2018. PMID: 29411205 Free PMC article.
-
Mapping the genomic landscape of inherited retinal disease genes prioritizes genes prone to coding and noncoding copy-number variations.Genet Med. 2018 Feb;20(2):202-213. doi: 10.1038/gim.2017.97. Epub 2017 Jul 27. Genet Med. 2018. PMID: 28749477 Free PMC article.
-
Conditional loss of Spata7 in photoreceptors causes progressive retinal degeneration in mice.Exp Eye Res. 2018 Jan;166:120-130. doi: 10.1016/j.exer.2017.10.015. Epub 2017 Oct 31. Exp Eye Res. 2018. PMID: 29100828 Free PMC article.
References
-
- den Hollander AI, Roepman R, Koenekoop RK, Cremers FP. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res. 2008;27:391–419. - PubMed
-
- Stone EM. Leber congenital amaurosis - a model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture. Am J Ophthalmol. 2007;144:791–811. - PubMed
-
- Heckenlively JR, Foxman SG, Parelhoff ES. Retinal dystrophy and macular coloboma. Doc Ophthalmol. 1988;68:257–71. - PubMed
-
- Perrault I, Rozet JM, Gerber S, Ghazi I, Leowski C, Ducroq D, Souied E, Dufier JL, Munnich A, Kaplan J. Leber congenital amaurosis. Mol Genet Metab. 1999;68:200–8. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
