

Mutations in DVL1 cause an osteosclerotic form of Robinow syndrome
- PMID: 25817014
- PMCID: PMC4385193
- DOI: 10.1016/j.ajhg.2015.02.010
Mutations in DVL1 cause an osteosclerotic form of Robinow syndrome
Abstract
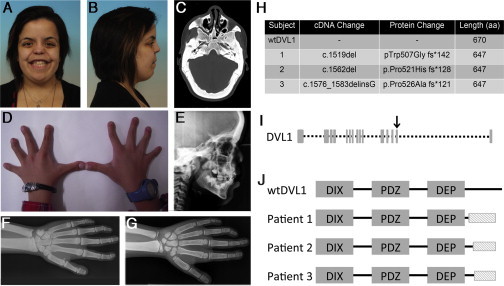
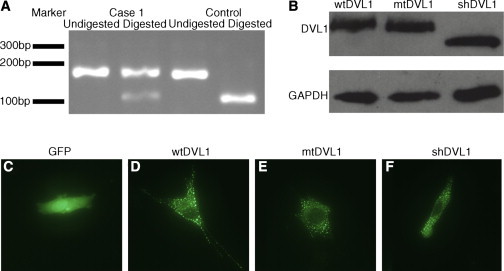
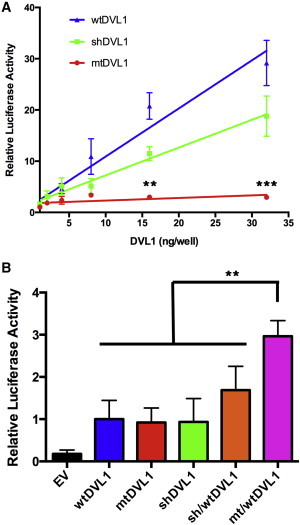
Robinow syndrome (RS) is a phenotypically and genetically heterogeneous condition that can be caused by mutations in genes encoding components of the non-canonical Wnt signaling pathway. In contrast, germline mutations that act to increase canonical Wnt signaling lead to distinctive osteosclerotic phenotypes. Here, we identified de novo frameshift mutations in DVL1, a mediator of both canonical and non-canonical Wnt signaling, as the cause of RS-OS, an RS subtype involving osteosclerosis, in three unrelated individuals. The mutations all delete the DVL1 C terminus and replace it, in each instance, with a novel, highly basic sequence. We showed the presence of mutant transcript in fibroblasts from one individual with RS-OS and demonstrated unimpaired protein stability with transfected GFP-tagged constructs bearing a frameshift mutation. In vitro TOPFlash assays, in apparent contradiction to the osteosclerotic phenotype, revealed that the mutant allele was less active than the wild-type allele in the canonical Wnt signaling pathway. However, when the mutant and wild-type alleles were co-expressed, canonical Wnt activity was 2-fold higher than that in the wild-type construct alone. This work establishes that DVL1 mutations cause a specific RS subtype, RS-OS, and that the osteosclerosis associated with this subtype might be the result of an interaction between the wild-type and mutant alleles and thus lead to elevated canonical Wnt signaling.
Copyright © 2015 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Mazzeu J.F., Pardono E., Vianna-Morgante A.M., Richieri-Costa A., Ae Kim C., Brunoni D., Martelli L., de Andrade C.E., Colin G., Otto P.A. Clinical characterization of autosomal dominant and recessive variants of Robinow syndrome. Am. J. Med. Genet. A. 2007;143:320–325. - PubMed
-
- Afzal A.R., Rajab A., Fenske C.D., Oldridge M., Elanko N., Ternes-Pereira E., Tüysüz B., Murday V.A., Patton M.A., Wilkie A.O., Jeffery S. Recessive Robinow syndrome, allelic to dominant brachydactyly type B, is caused by mutation of ROR2. Nat. Genet. 2000;25:419–422. - PubMed
-
- van Bokhoven H., Celli J., Kayserili H., van Beusekom E., Balci S., Brussel W., Skovby F., Kerr B., Percin E.F., Akarsu N., Brunner H.G. Mutation of the gene encoding the ROR2 tyrosine kinase causes autosomal recessive Robinow syndrome. Nat. Genet. 2000;25:423–426. - PubMed
-
- Niehrs C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012;13:767–779. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
