

Loss-of-function alanyl-tRNA synthetase mutations cause an autosomal-recessive early-onset epileptic encephalopathy with persistent myelination defect
- PMID: 25817015
- PMCID: PMC4385183
- DOI: 10.1016/j.ajhg.2015.02.012
Loss-of-function alanyl-tRNA synthetase mutations cause an autosomal-recessive early-onset epileptic encephalopathy with persistent myelination defect
Abstract
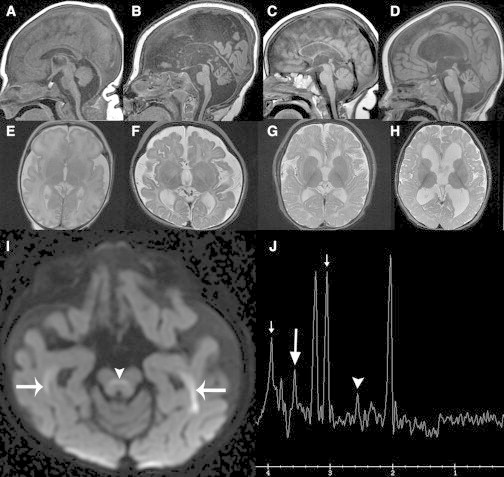
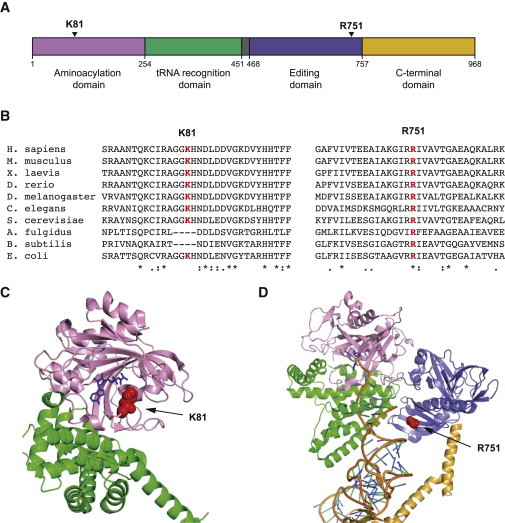
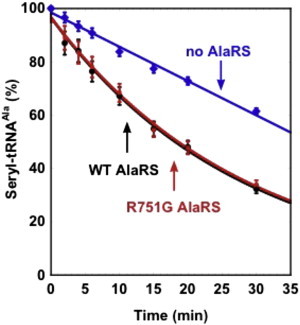
Mutations in genes encoding aminoacyl-tRNA synthetases are known to cause leukodystrophies and genetic leukoencephalopathies-heritable disorders that result in white matter abnormalities in the central nervous system. Here we report three individuals (two siblings and an unrelated individual) with severe infantile epileptic encephalopathy, clubfoot, absent deep tendon reflexes, extrapyramidal symptoms, and persistently deficient myelination on MRI. Analysis by whole exome sequencing identified mutations in the nuclear-encoded alanyl-tRNA synthetase (AARS) in these two unrelated families: the two affected siblings are compound heterozygous for p.Lys81Thr and p.Arg751Gly AARS, and the single affected child is homozygous for p.Arg751Gly AARS. The two identified mutations were found to result in a significant reduction in function. Mutations in AARS were previously associated with an autosomal-dominant inherited form of axonal neuropathy, Charcot-Marie-Tooth disease type 2N (CMT2N). The autosomal-recessive AARS mutations identified in the individuals described here, however, cause a severe infantile epileptic encephalopathy with a central myelin defect and peripheral neuropathy, demonstrating that defects of alanyl-tRNA charging can result in a wide spectrum of disease manifestations.
Copyright © 2015 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Almalki A., Alston C.L., Parker A., Simonic I., Mehta S.G., He L., Reza M., Oliveira J.M.A., Lightowlers R.N., McFarland R. Mutation of the human mitochondrial phenylalanine-tRNA synthetase causes infantile-onset epilepsy and cytochrome c oxidase deficiency. Biochim. Biophys. Acta. 2014;1842:56–64. - PMC - PubMed
-
- Steenweg M.E., Ghezzi D., Haack T., Abbink T.E., Martinelli D., van Berkel C.G., Bley A., Diogo L., Grillo E., Te Water Naudé J. Leukoencephalopathy with thalamus and brainstem involvement and high lactate ‘LTBL’ caused by EARS2 mutations. Brain. 2012;135:1387–1394. - PubMed
-
- Yamashita S., Miyake N., Matsumoto N., Osaka H., Iai M., Aida N., Tanaka Y. Neuropathology of leukoencephalopathy with brainstem and spinal cord involvement and high lactate caused by a homozygous mutation of DARS2. Brain Dev. 2013;35:312–316. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
